
There has been a growing appreciation in recent years that gene function is frequently context-dependent, with a large part of that context provided by the activities of other genes. The Gillis lab focuses on characterizing these shared patterns of gene activity through co-expression networks and showing how they can lead to changes in cell function, particularly in single cell expression data.
A dominant interest within computational biology is the analysis of gene networks to provide insight into diverse levels of functional activity, typically starting with regulatory interactions and moving up to more diffuse associations important for understanding systemic dynamics. But trying to understand how genes interact to produce function is a hugely complicated problem and one that appears likely to become more so as genomic information becomes more detailed. Historically, many attempts to understand gene function through networks have leveraged a biological principle known as “guilt by association.” It suggests that genes with related functions tend to share properties (e.g., physical interactions). In the past decade, this approach has been scaled up for application to large gene networks, becoming a favored way to grapple with the complex interdependencies of gene functions in the face of floods of genomics and proteomics data. Gillis’ work centers on identifying the limits of the approach and making fundamental improvements to its operation, as well as applying those improvements to understanding cell biology.

The CSHL School of Biological Sciences’ class of 2025
May 5, 2025
The School of Biological Sciences awarded Ph.D. degrees to nine students this year. Read some of their stories and reflections on their time at CSHL.

The CSHL School of Biological Sciences’ class of 2024
May 5, 2024
The School of Biological Sciences awarded Ph.D. degrees to 11 students this year. Here are some stories and reflections from their time at CSHL.

Cold Spring Harbor Laboratory 2022 Ph.D.’s
May 1, 2022
The School of Biological Sciences awarded Ph.D. degrees to ten students this year. Here are some stories and memories from their time at CSHL.

Building on 150 years of neuroanatomy
October 7, 2021
Learn more about how researchers reached a milestone in a years-long effort to catalog the cells of the human, mouse, and monkey brains.
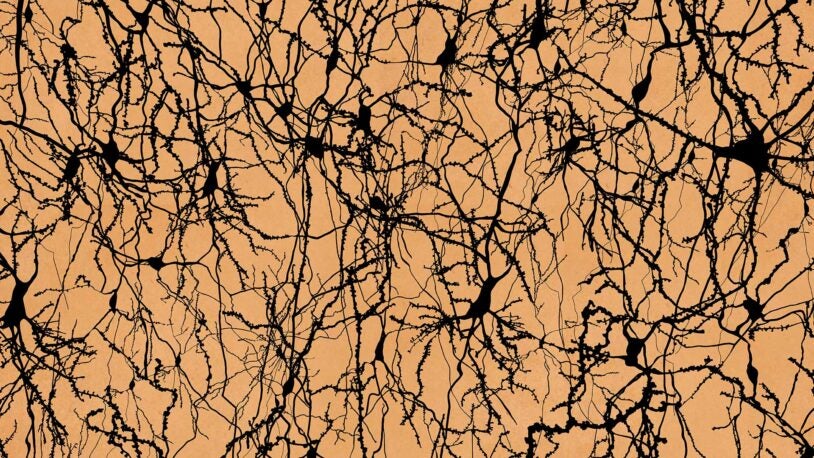
Think a census of humans is hard? Try counting their brain cells!
October 6, 2021
CSHL researchers and other collaborators reached a milestone in a years-long effort to catalog the cells of the human, mouse, and monkey brains.

URP: Summer camp for undergrads
July 29, 2021
The Undergraduate Research Program brings college students from around the world to CSHL for a summer of research and fun.
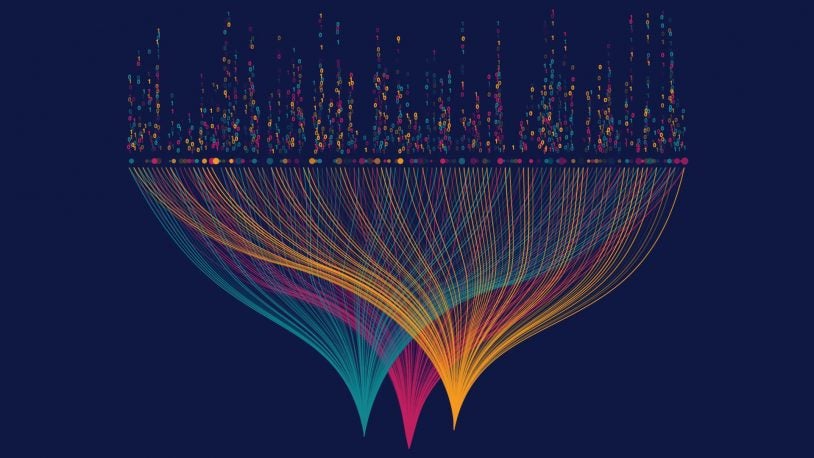
Using “guilt by association” to classify cells
July 14, 2021
Using a new computational statistics tool, CSHL researchers classify cells to understand how an organism functions.

Solving genetic disease puzzles with quantitative biology
June 17, 2021
CSHL quantitative biologist Jesse Gillis teams up with an immunology specialist at Northwell Health to analyze a complex genetic disorder.
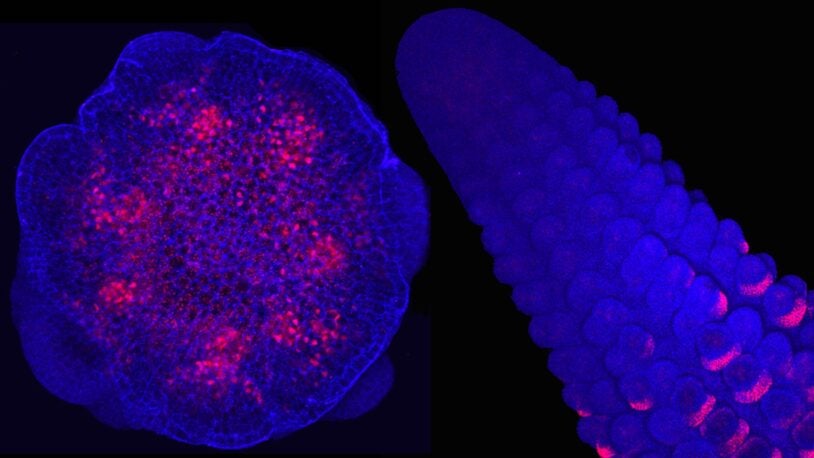
Building a corn cob—cell by cell, gene by gene
January 26, 2021
CSHL scientists are piecing together the genes that control how corn develops.

NIH grant awarded for interneuron research
April 4, 2019
CSHL postdoc Maggie Crow will use her NIH grant to pursue the quantification and analysis of specific types of neurons in the brain.