Cold Spring Harbor Laboratory is an HHMI designated Pacific Biosciences (PacBio) sequencing site. We provide PacBio sequencing services to all HHMI investigators located at nearby non-profit research institutions.
We also provide fee-for-service sequencing to non-HHMI investigators around the world. Our users includes scientists from the US, UK, Italy, France, Belgium, Russia, South Africa, New Zealand and Australia
Explore all the CSHL Core Facilities
Contact:
Sara Goodwin
sgoodwin@cshl.edu
516-422-4086
 Sequencing Service:
Sequencing Service:
We offer full service sequencing on the Pacific Biosciences RS instrument. We will QC the DNA, make the PacBio sequencing library and sequence the library on the appropriate number of SMRT cells.
Furthermore, we will help you design your sequencing experiment (insert size, type of run, number of SMRT cells, etc.) to help ensure that you will receive the data you need for your research.
Final Data:
Because PacBio sequencing libraries are made from double stranded DNA molecules with hairpin loops (adapters) ligated at both ends, the final product is a circular molecule. Therefore, the polymerase can sequence both strand of the insert multiple times, separated by the adapter sequences. We remove the adapter sequences, breaking up the polymerase reads into subreads. If the polymerase read did not reach the end of the insert, then you get one subread = polymerase read. If the polymerase reach the end of the insert, go through the circular adapter and start sequencing the opposite strand, then you get two subreads. If the polymerase goes N times around the circular molecule, then you get N subreads from one polymerase read. A read_of_insert (aka CCS or HiFi read) is the consensus sequence obtained by doing a multi-sequence alignment using all the subreads coming from one polymerase read.
The data are provided on our SFTP server or can be accessed via a user provided cloud service.
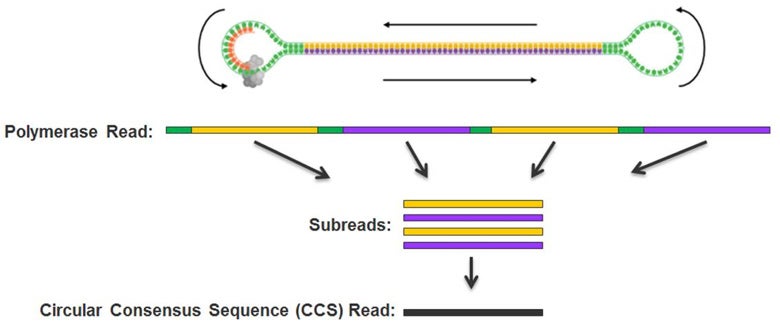
PacBio technology revealed:
- http://www.pacb.com/products/smrt-technology
- http://www.youtube.com/watch?v=_B_cUZ8hSYU
- http://www.pacificbiosciences.com/applications/overview/
Current performance of the Pacific Biosciences Sequel II sequencer:
A typical SMRT Cell will generate between 4,000,000 and 6,000,000 reads. The average polymerase read length for a HiFi run is 12kb. The average polymerase read length for a Continuous Long Read (CLR) run is 20kb. Subread N50 sequence lengths are as high as 30Kb for large insert libraries.
A HiFi SMRT cell typically yields between 20 and 30 billion unique >Q20 bases. A CLR SMRT cell typically yields between 50 and 90 billion bases when sequencing a 30kb genomic library.
SMRT cell yield is very dependent on DNA quality, especially the number of single strand DNA nicks.
PCR amplicon libraries, including cDNA libraries have lower total yield than genomic libraries, though the number of unique reads in the 3-5 million reads range. Due to the impact of single strand nicks and other artifact, care must be taken when generating amplicons >5kb. Please reach out to the core for guidelines on generating compatible fragments. SMRT cell average yield is above 1,000 million bases, and our best yield is 1,818 million bases per cell.
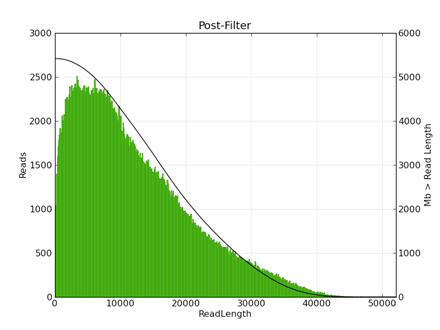
The following figures show typical polymerase read length (fig 1) and subread length (fig 2) histograms for a 20kb library size selected on the BluePippin (15-50kb) ) and sequenced using the P6 enzyme on 8 SMRT cells (680Mb/SMRT cell).
 |
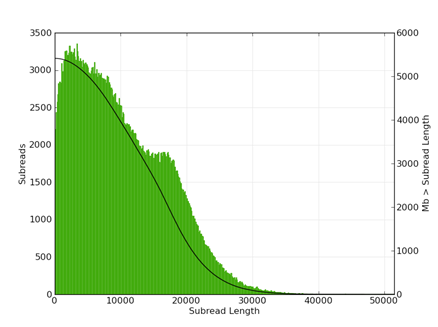 |
| Figure 1 | Figure 2 |
The error rates are as advertised by the company, about 12% with a single pass over a template. Hence, in the long read mode, your DNA molecule would be sequenced a single time with this overall error rate. In the circular consensus mode (CCS), a molecule would be sequenced several times, and the consensus sequence (CCS) error rate will be considerably lower (exact error rate depends on the number of individual sequences used to generate the CCS sequence).
Details of the full range of applications can be found at the PacBio applications page; http://www.pacb.com/applications/overview/index.html
For understanding accuracy in SMRT sequencing, see the following: Understanding Accuracy SMRT Sequencing
We are a Certified PacBio service provider. We offer sequencing to both internal and external users, including for profit organizations. HHMI investigators are entitled to internal pricing. Please reach out to the DNA Sequencing Shared Resource core (nextgenseq@cshl.edu) for pricing and consultation.