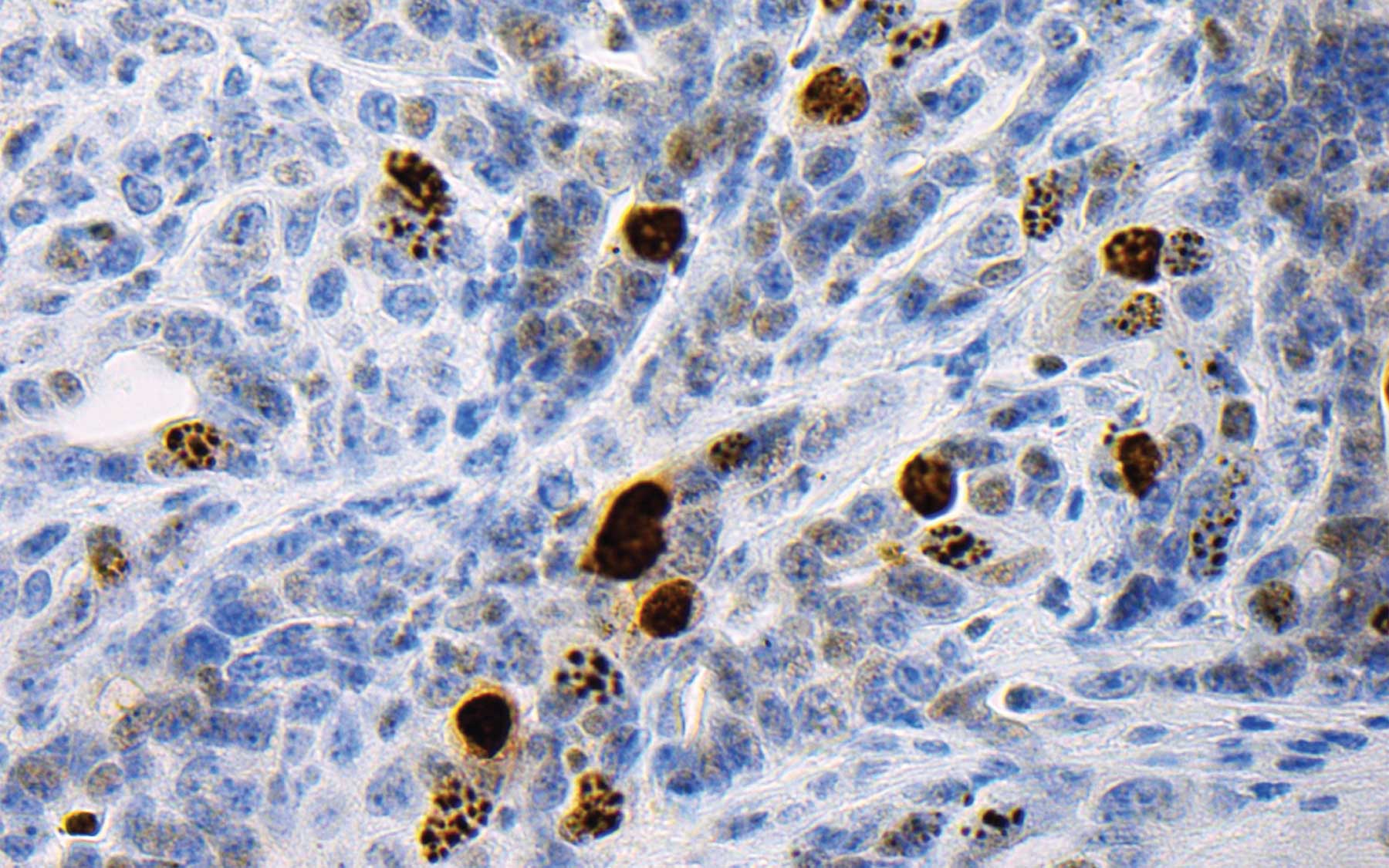
A strategy based on mimicking the suppression of antioxidant-promoting NRF2
Cold Spring Harbor, NY — Reducing levels of antioxidants in pancreatic cancer cells can help kill them, newly published research reveals, suggesting an entirely new treatment strategy for the notoriously lethal illness, in which less than 5 percent of patients survive 5 years.
Although it has become almost a matter of conventional wisdom in popular culture that raising antioxidant levels in the body tends to keep cancer at bay, a team at Cold Spring Harbor Laboratory (CSHL) demonstrates in a series of sophisticated experiments that in the specific context of pancreatic cells on the road to cancer or already in a malignant state, the last thing one wants to do is to raise antioxidant levels.
Oxidizing and anti-oxidizing agents are created in every cell, and are kept in a very precise balance when cells are healthy. The CSHL team, led by Professor David Tuveson, M.D., Ph.D., who is also director of research for The Lustgarten Foundation, proceeds from the insight that proliferating cancer cells present a special case: more oxidants are being made in malignant cells, but more anti-oxidants are being made, too, countering the impact of rising oxidation. Without commensurately more anti-oxidants, malignant cells will die due to excessive oxidation.
“Of course, that’s exactly what we want cancer cells to do—to burn themselves out,” observes Iok In Christine Chio, a postdoctoral investigator in the Tuveson lab who led experiments reported today in the journal Cell. “The therapeutic principle our lab is testing is whether, by increasing the level of oxidation in cancer cells, we can cause pre-malignant and malignant cells to die.”
Most existing forms of cancer therapy, including ionizing radiation treatments and chemotherapy, depend on the killing power of oxidation. Anti-oxidants do indeed have a role in cellular health; but when it comes to killing cancer cells, they are anathema.
When cells detect excessive oxidation, they literally commit suicide, following a built-in program called apoptosis. One way of increasing oxidation in cancer cells is to decrease levels of antioxidants in those same cells. A question addressed by Tuveson’s team was how best to do this—and do it in a way that does not harm healthy cells. They focused on a protein called NRF2 (pronounced “nerf-2”). Scientists call it a master regulator of redox homeostasis—by which they mean that it’s one of the switches one can tweak to disturb the exquisite balance between oxidation and reduction in cancer cells.
When NRF2 is active, cells synthesize a chemical called glutathione, an important antioxidant. It makes sense, then, to try to reduce NRF2 activity or knock it out of action altogether. This is not possible, for two reasons. One is that it is a transcription factor—a protein that regulates the activity of other genes. Transcription factors are famously difficult to target with drugs. But “you probably wouldn’t want to knock it out, in any case,” says Chio, “because in addition to promoting production of glutathione, NRF2 has a role in regulating several hundred different genes.” One can’t delete it from a cell without impacting many other processes.
Tuveson, Chio and other members of the team arrived at a different strategy after conducting experiments in pancreas cells grown in the lab and testing a number of new therapeutic approaches in an animal model of pancreatic cancer.
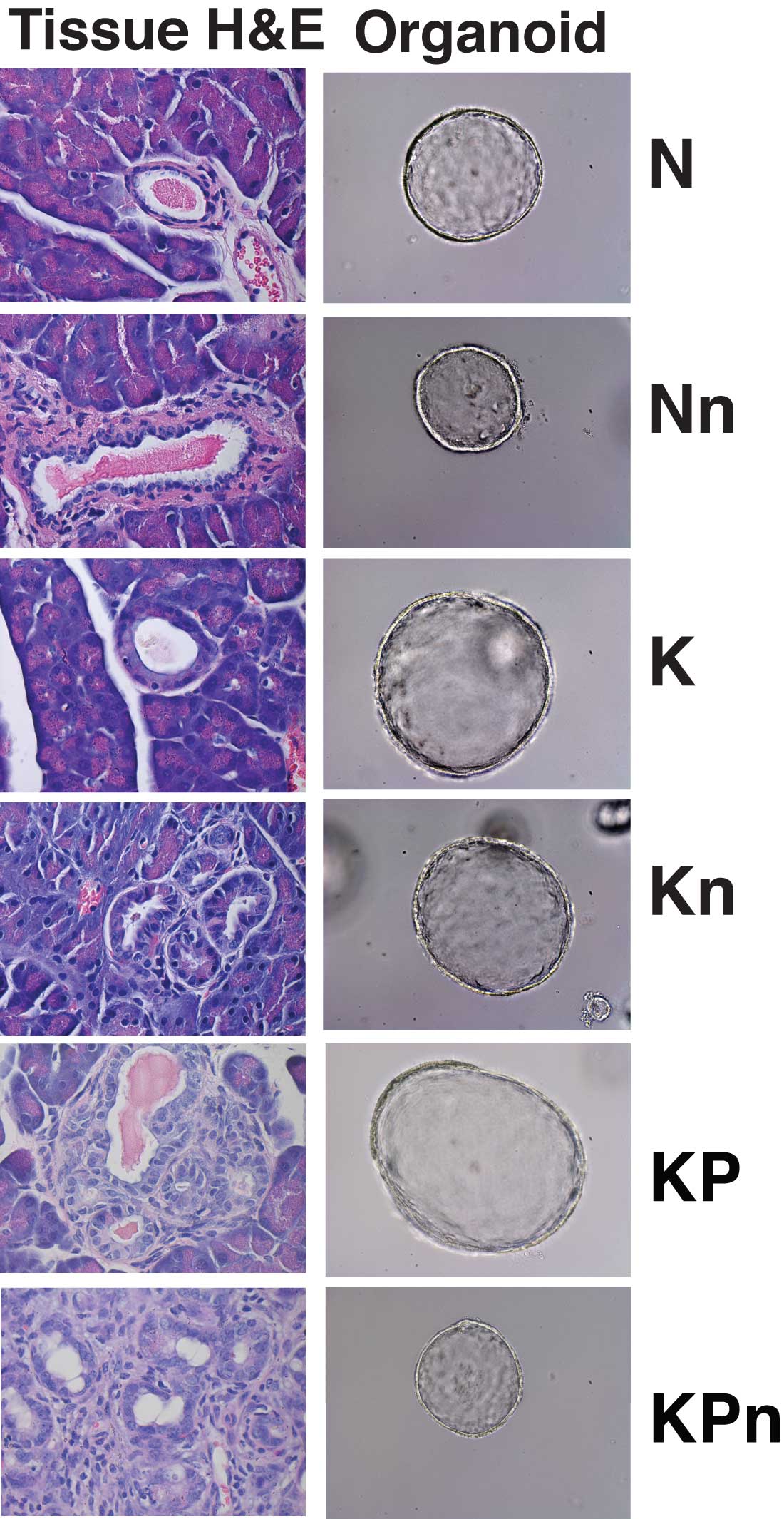
Using a panel of pancreas organoids—spherical agglomerations of pancreas cells sampled from people with pancreatic cancer and from the healthy pancreas—they were able to observe what happens when NRF2 is completely eliminated. The test was run in normal, pre-malignant and malignant pancreas organoids. The pre-malignant organoids carried cellular mutations in the kras gene, which is aberrant in nearly all human pancreatic cancers. The malignant organoids bore that mutation plus a mutation that inactivated the powerful tumor suppressor gene p53. That mutation along with kras is seen in most human malignancies.
These experiments provided a critical clue: The team saw that when NRF2 is missing, the machinery in cells that translates messages from genes into proteins—the process of protein synthesis—is very sensitive to fluctuations in the balance between oxidants and anti-oxidants. Crucially, however, protein synthesis was not impacted in normal pancreas cells. “We were very excited when we saw this,” Chio says. “This meant that if we could find a way of reducing antioxidants, protein synthesis would only be impacted in precancerous and malignant cells, a potentially powerful therapeutic strategy.”
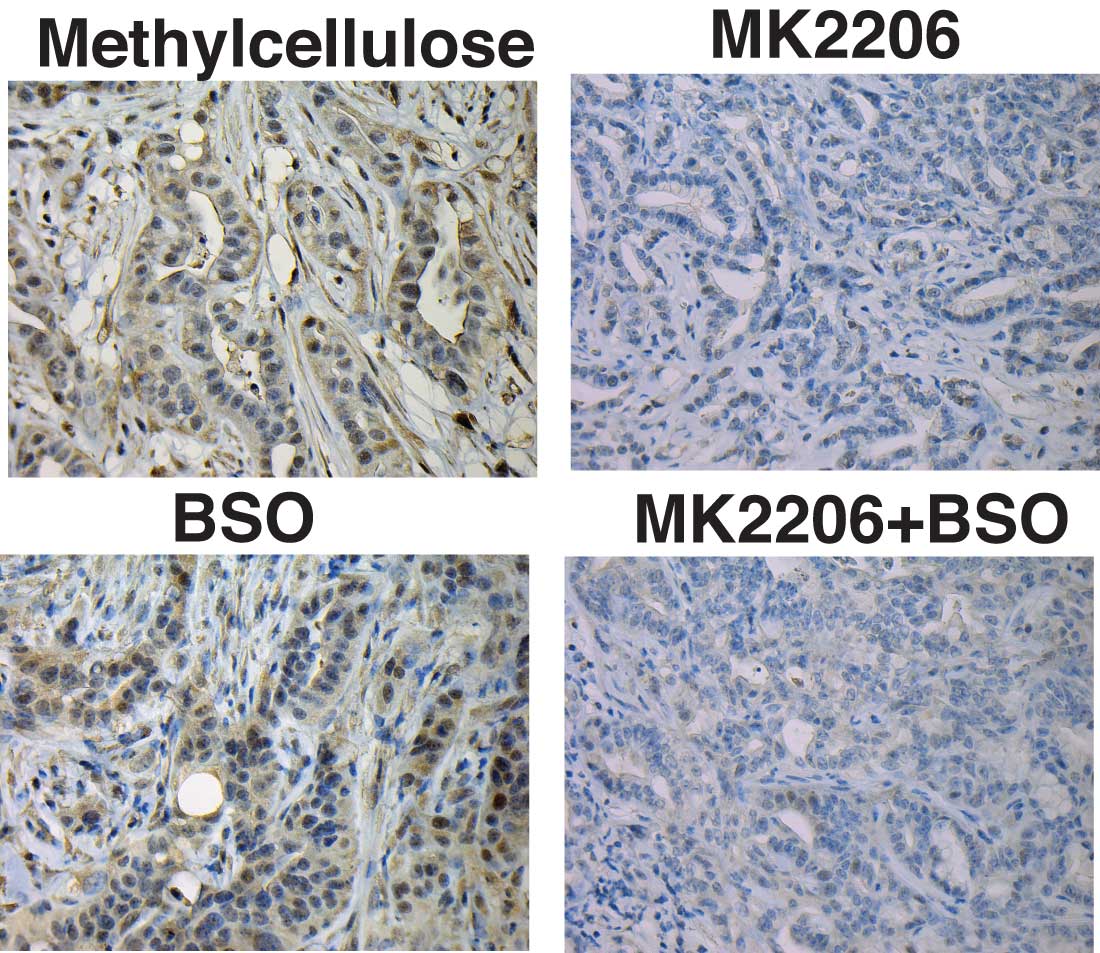
This phenomenon is called synthetic lethality. It occurs when a condition affecting both healthy and unhealthy cells is lethal only to the unhealthy cells.
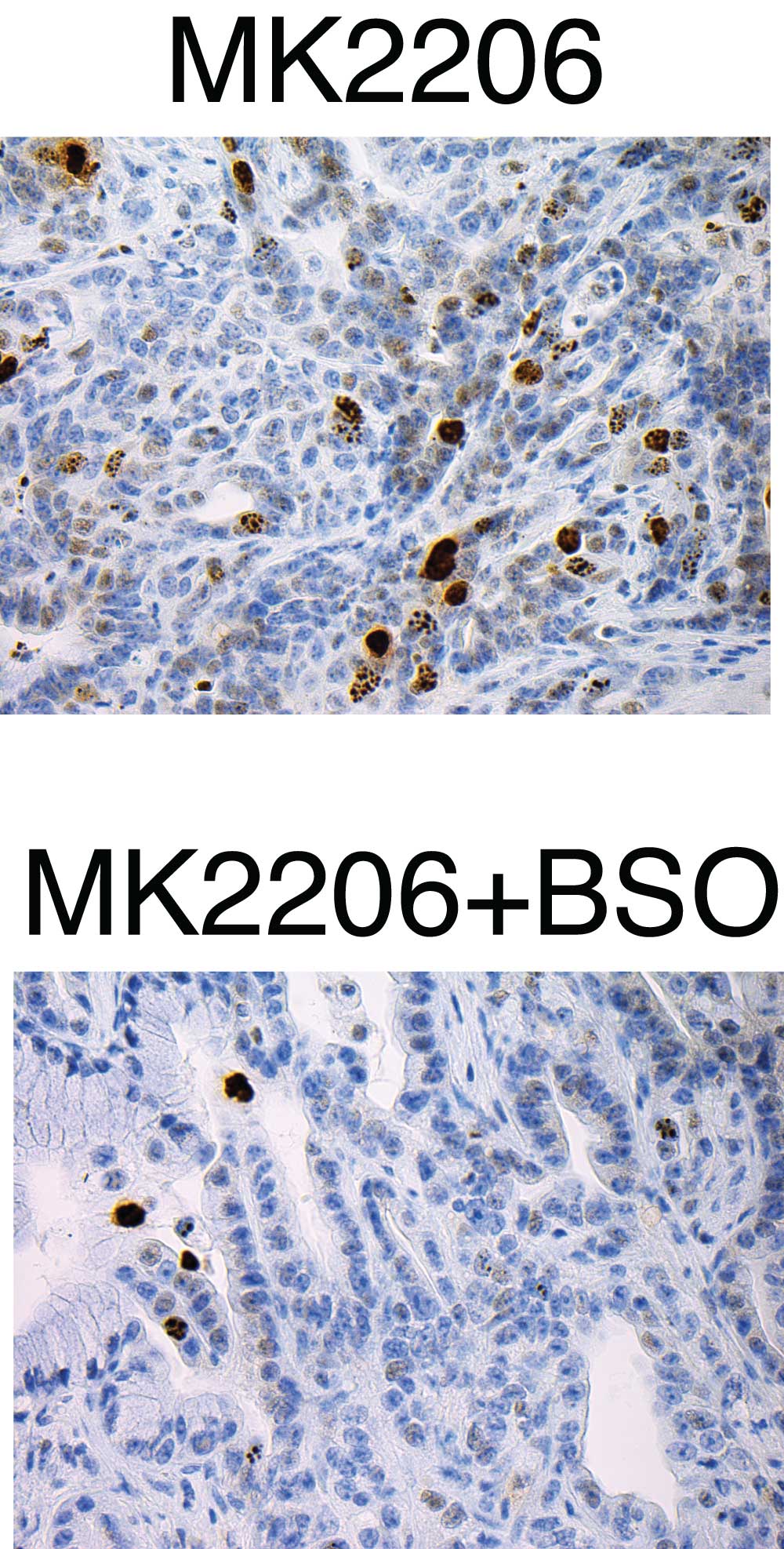
The team came up with a two-drug cocktail, which they delivered to mice that model pancreatic cancer. One of the agents, called an AKT inhibitor, is designed to inhibit the beginning of translation process that leads to protein synthesis. The other agent, called BSO, inhibits the synthesis of glutathione, an antioxidant.
AKT inhibitors, administered alone, have produced only modest survival benefits in both mouse models and human patients. “We hoped that by combining them with BSO we might generate a synergy that would boost its effectiveness,” says Chio. In effect, the strategy was to mimic biochemically what would happen if they were able to knock out or reduce levels of NRF2 in cancer cells. It was the rationale for hitching BSO to the AKT inhibitor.
“This is where our pancreas organoid system was so valuable,” Chio says. “We were able to test this idea and see that this approach was synthetically lethal—it did increase the killing power of the AKT inhibitor, but the synergy was not present in the setting of normal pancreas cells,” she says. In other words, normal pancreas cells were not harmed by the treatment.
The team’s new paper reports a synergistic effect in the mouse model of pancreas cancer, as well, proving the concept of killing more cancer by decreasing cancer cell antioxidant levels. Not fully satisfied with the survival benefit noted—comparatively small in the mice—they believe they can optimize the treatment by combining different translation inhibitors and repressors of antioxidant synthesis. This work is well under way.
The approach is potentially pertinent in other cancers in which kras mutations place cells on the road to malignancy. These include certain types of lung cancer and breast cancer.
Dr. Tuveson, who is director of The Lustgarten Foundation Pancreatic Cancer Research Laboratory as well as director of research for The Lustgarten Foundation and Deputy Director of the CSHL Cancer Center, says he is excited about the research.
“Pancreatic cancer employs NRF2 as one of its henchmen to promote bad behavior, but the details were previously unclear,” Dr. Tuveson says. “By developing organoid models, Christine, proteomics expert Darryl Pappin of CSHL and their collaborators pursued the basic biology of NRF2 to unravel the molecular underpinnings of this protein and thereby propose new therapeutic approaches for patients. We plan to translate Christine’s findings to early phase clinical trials in the near future.”
Written by: Peter Tarr, Senior Science Writer | publicaffairs@cshl.edu | 516-367-8455
Funding
The research discussed in this release was directly supported by: Cancer Center Support Grant 5P30CA045508; The Lustgarten Foundation; Cold Spring Harbor Laboratory Association; National Institutes of Health (5P30CA45508-26, 5P50CA101955-07, 1U10CA180944-01, 5U01CA168409-3, and 1R01CA190092-01); Carcinoid Foundation, PCUK, and the David Rubinstein Center for Pancreatic Cancer Research at MSKCC.
Citation
“NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer” appears online in Cell July 28, 2016. The authors are: Iok In Christine Chio, Seyed Mehdi Jafarnejad, Mariano Ponz-Sarvise, Youngkyu Park, Keith Rivera, Wilhelm Palm, John Wilson, Vineet Sangar, Yuan Hao, Daniel Öhlund, Kevin Wright, Dea Filippini, Eun Jung Lee, Brandon Da Silva, Christina Schoepfer, John Erby Wilkinson, Jonathan Buscaglia, Gina M. DeNicola, Herve Tiriac, Molly Hammell, Howard C. Crawford, Edward E. Schmidt, Craig B. Thompson, Darryl J. Pappin, Nahum Sonenberg and David A. Tuveson. The paper can be accessed at: http://www.cell.com/cell/home
Principal Investigator

David Tuveson
Professor
Roy J. Zuckerberg Professor of Cancer Research
Cancer Center Director
M.D., Ph.D., Johns Hopkins University, 1994