Online app could help clinicians choose the best treatments by comparing genetic fingerprints of individual cells
Cold Spring Harbor, NY — Seemingly similar cells often have significantly different genomes. This is often true of cancer cells, for example, which may differ one from another even within a small tumor sample, as genetic mutations within the cells spread in staccato-like bursts. Detailed knowledge of these mutations, called copy number variations, in individual cells can point to specific treatment regimens.
The problem is that current techniques for acquiring this knowledge are difficult and produce unreliable results. Today, scientists at Cold Spring Harbor Laboratory (CSHL) publish a new interactive analysis program called Gingko that reduces the uncertainty of single-cell analysis and provides a simple way to visualize patterns in copy number mutations across populations of cells.
The open-source software, which is freely available online, will improve scientists’ ability to study this important type of genetic anomaly and could help clinicians better target medications based on cells’ specific mutation profiles. The software is described online today in Nature Methods.
Mutations come in many forms. For example, in the most common type of mutation, variations may exist among individual people—or cells—at a single position in a DNA sequence. Another common mutation is a copy number variation (CNV), in which large chunks of DNA are either deleted from or added to the genome. When there are too many or too few copies of a given gene or genes, due to CNVs, disease can occur. Such mutations have been linked not only with cancer but a host of other illnesses, including autism and schizophrenia.
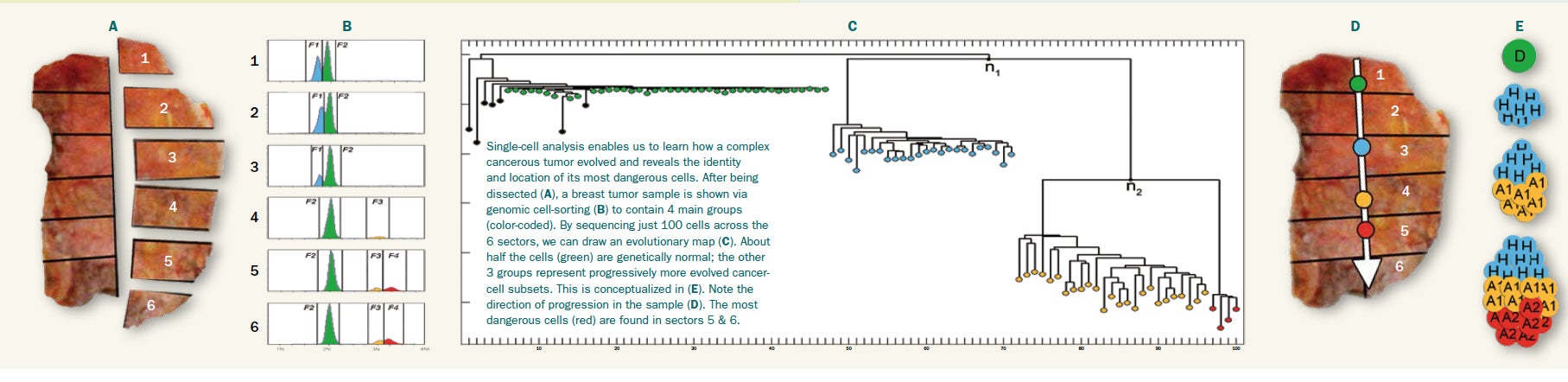
Researchers can learn a lot by analyzing CNVs in bulk samples—from a tumor biopsy, for example—but they can learn more by investigating CNVs in individual cells. “You may think that every cell in a tumor would be the same, but that’s actually not the case,” says CSHL Associate Professor Michael Schatz.
“We’re realizing that there can be a lot of changes inside even a single tumor,” says Schatz. “If you’re going to treat cancer, you need to diagnose exactly what subclass of cancer you have.” Simultaneously employing different drugs to target different cancer subclasses could prevent remission, scientists have proposed.
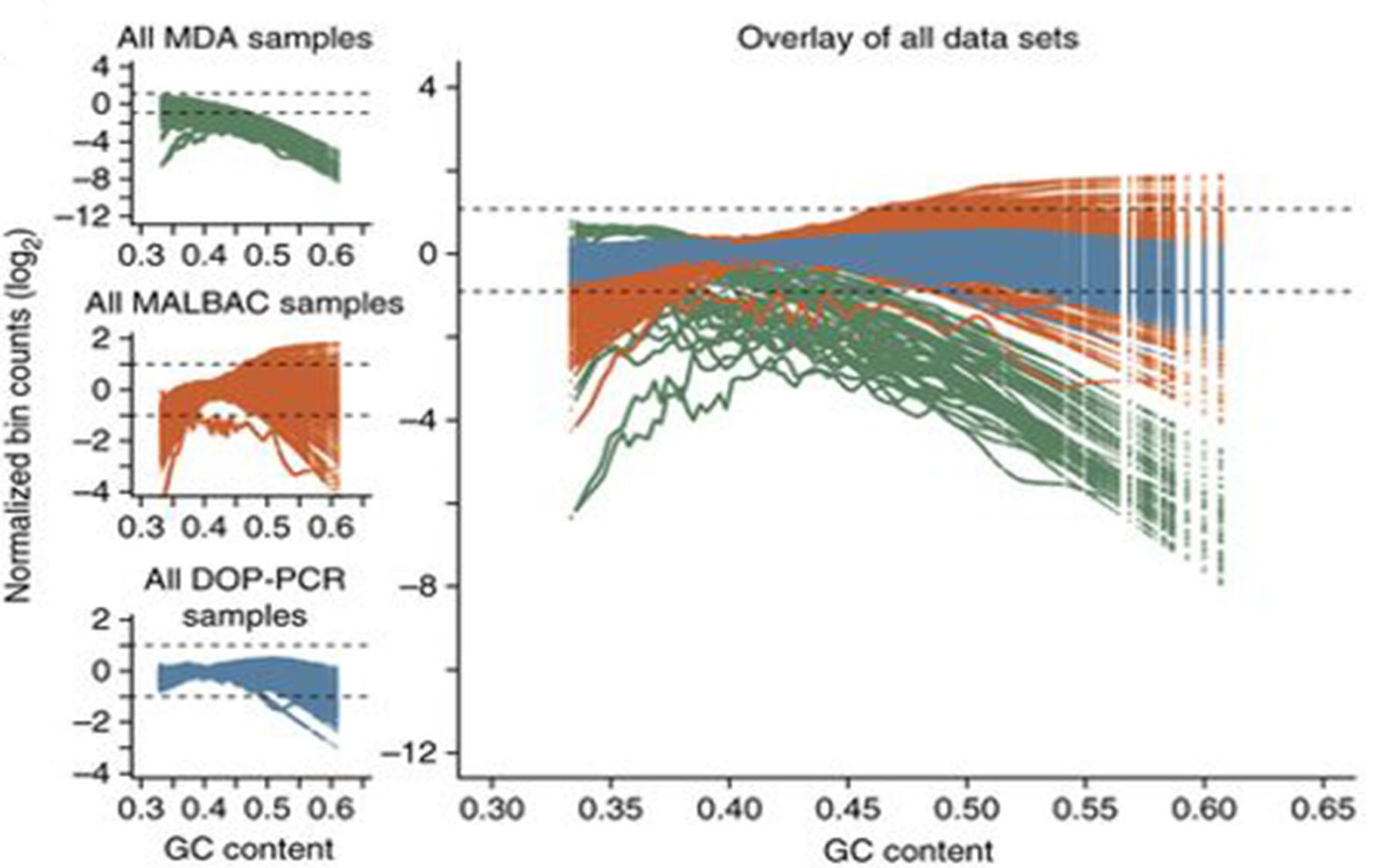
One powerful single-cell analytic technique for exploring CNV is whole genome sequencing. The challenge is that, before sequencing can be done, the cell’s DNA has to be amplified many times over. This process is rife with errors, with some arbitrary chunks of DNA being amplified more than others. In addition, because many labs use their own software to examine CNVs, there is little consistency in how researchers analyze their results.
To address these two challenges, Schatz and his colleagues created Gingko. The interactive, web-based program automatically processes sequence data, maps the sequences to a reference genome, and creates CNV profiles for every cell that can then be viewed with a user-friendly graphical interface. In addition, Gingko constructs phylogenetic trees based on the profiles, allowing cells with similar copy number mutations to be grouped together.
Importantly, Gingko, which Schatz and his colleagues validated by reproducing the findings of five major single-cell studies, also analyzes patterns in the sequence reads in order to recognize, and greatly reduce, amplification errors.
Schatz and his team named their software after the gingko tree, which has many well-documented therapeutic benefits. “We like to think our Gingko ‘trees’ will provide benefits as well,” says Schatz, referring to the graphical way that CNV changes are represented by analysts. Right now, CNV is not a commonly used diagnostic measurement in the clinic. “We’re looking into the best way of collecting samples, analyzing them, and informing clinicians about the results,” says Schatz. He adds that CSHL has collaborations with many hospitals, notably Memorial Sloan Kettering Cancer Center and the North Shore-LIJ Health System, to bring single-cell analysis to the clinic.
For Schatz, Gingko represents a culmination of CSHL’s efforts over the past decade—spearheaded by CSHL Professor Michael Wigler—to pioneer techniques for studying single cells. “Cold Spring Harbor has established itself as the world leader in single-cell analysis,” says Schatz. “We’ve invented many of the technologies and techniques important to the field and now we’ve taken all this knowledge and bundled it up so that researchers around the world can take advantage of our expertise.”
Written by: Chris Palmer, Science Writer | publicaffairs@cshl.edu | 516-367-8455
Funding
The research described in the release was supported by the National Institutes of Health (NIH), the National Science Foundation, the Starr Cancer Consortium, the Breast Cancer Research Foundation, the Simons Foundation, the Susan G. Komen Foundation, the Prostate Cancer Foundation, the Cold Spring Harbor Laboratory (CSHL) Cancer Center and the Watson School of Biological Sciences at CSHL through an NIH training grant.
Citation
“Interactive analysis and assessment of single-cell copy-number variations,” appears online September 7, 2015 in Nature Methods. The authors are Tyler Garvin, Robert Aboukhalil, Jude Kendall, Timour Baslan, Gurinder S. Atwal, James Hicks, Michael Wigler and Michael C. Schatz. The paper can be viewed at: http://www.nature.com/nmeth/journal/vaop/ncurrent/full/nmeth.3578.html
Principal Investigator

Michael Schatz
Adjunct Associate Professor