Genetic causation of ASD appears to be highly diverse; thoughts on why fewer girls have autism
Cold Spring Harbor, NY — A clinically extensive and mathematically powerful study of 1000 families with one autistic child and one unaffected sibling has validated a controversial theory of autism’s complex genetic causation. The study for the first time estimates the minimum number of locations in the human genome—250 to 300—where gene copy number variation (CNV) can give rise to autism spectrum disorder (ASD). It also sheds new light on the long observed but little understood “gender bias” of autism, an illness that typically manifests by age 3 and affects about four times more boys than girls.
The study, along with an accompanying paper exploring the possible brain and neuronal pathologies to which the observed CNVs and other genetic anomalies may give rise, appears June 9 in the journal Neuron. The work was led by Professor Michael Wigler of Cold Spring Harbor Laboratory (CHSL), a pioneer in the analysis of genomes and a developer of key technologies that have made such analysis possible, in collaboration with Dennis Vitkup of Columbia University; Dan Levy, Michael Ronemus, Ivan Iossifov and Sarah Gilman of CSHL; and others.
“The causes of autism when fully fleshed out are likely to be very diverse,” Wigler says, “some of which may be treatable much more readily than others. However, the diversity of causes implies that an effective future treatment for one form of ASD may be specific only for a narrow subset of those affected.”
Four years ago, Wigler and colleagues began to publish findings about the genetics of autism that surprised many experts in the field. Among other things, they noted the prevalence of “de novo” genetic mutations in affected children. These are mutations that did not appear in either parent and hence must have arisen spontaneously. In 2008, Wigler proposed that such cases, characterized by changes in gene copy number—duplicated or deleted genome segments that leave an individual with extra or missing copies of one or more genes—likely account for at least half of ASD cases.
There is little doubt that certain genetic mutations carried by an unaffected parent can be transmitted to a child and cause the illness. Wigler, however, proposed a “unified theory” of autism genetics that suggested such cases only account for about 25% of the total.
Rare and ultra-rare CNVs—and their possible biological consequences
The new study affirms the team’s earlier data on the relative importance of non-inherited, spontaneously occurring CNVs as likely causal factors in autism. Using a newly assembled sample population called the Simons Simplex Collection which included about 1000 families, consisting of both birth parents and in most cases two children, one with ASD and the other unaffected, Wigler and colleagues saw the earlier-observed effect, only “this time on a much larger scale.” The new sample collection is deemed very important by the research team. It intentionally excludes families with more than one child with ASD (“multiplex families”) since such cases are almost always inherited. A sample composed only of “simplex” families makes possible a much more accurate comparison of the relative frequency of spontaneous and inherited causal mutations.
Using a refined version of a technique called comparative genomic hybridization, but using detection devices called microarrays that were about four times as powerful as those used in prior studies, along with a suite of powerful quantitative analysis tools, the team was able to consistently resolve much smaller genomic irregularities than previously possible. This revealed a significantly greater number of genome areas where spontaneous DNA deletions and duplications “hit” genes thought to contribute substantially to ASD.
The team estimates a minimum of 250 to 300 places in the genome where CNVs give rise to ASD. Most of the CNVs detected were only seen once and are classified, individually, as rare. This is a potential source of confusion since these rare events, taken in total, are the source of at least half of all autism, if Wigler’s theory is correct.
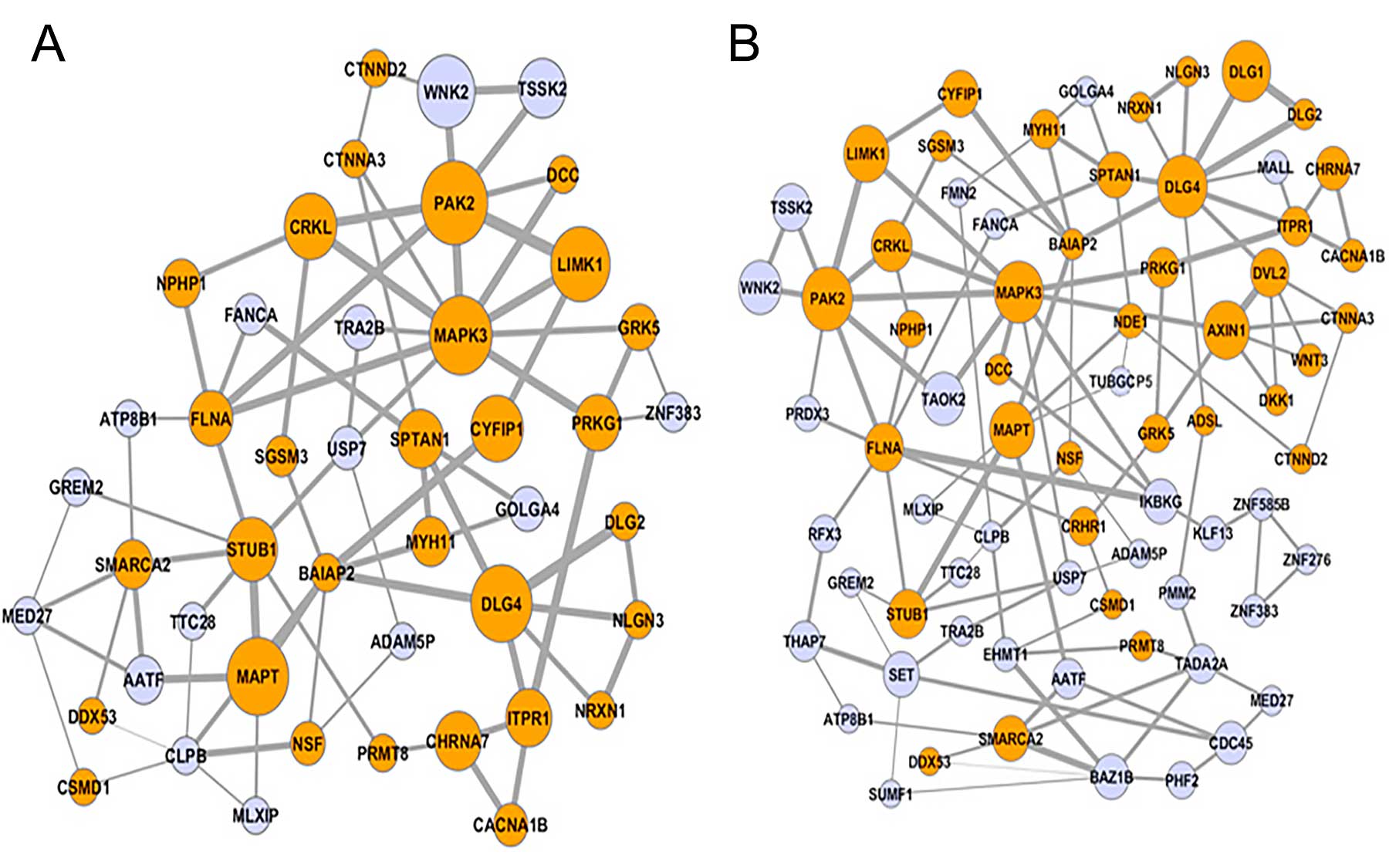
As the team notes in their paper, it is still very difficult to make the connection between the areas that the de novo CNVs hit and the genes these areas harbor. Nor is it clear what role those genes have in perturbing the biology of the developing brain, giving rise to autism. In the associated paper, Vitkup, Iossifov and others use a novel form of analysis called NETBAG (network-based analysis of genetic associations) to identify “the large biological network of genes affected by rare de novo CNVs in autism.” They note that this network is strongly related to genes previously implicated in studies of autism and intellectual disability. And they specify that the genes in question relate primarily to the development of synapses, the junctions between brain cells which are their transmission nodes; as well as the targeting of axonal fibers and the ability of young neurons to migrate. All are essential aspects of brain development.
Explaining gender skew and apparent female ‘resistance’ to autism
Subsequent sequence-based studies should be able to further resolve individual genes within the CNV regions, and significantly expand the list of candidate genes for autism. Even now, however, the data on spontaneous CNVs and a separate but also an important class of “ultra-rare” CNVs that were inherited by some of the affected children in the sample, reveal interesting and in some cases vexing “asymmetries,” the team reported.
Among these asymmetries: spontaneous causal CNVs were found in 8% of the affected children in the sample; this is four times their rate of occurrence in unaffected siblings. Since CNVs occur with equal frequency across the human population—we all have them, but in most of us the missing or extra genetic material doesn’t hit essential genes or contribute to illness—the fact that they are seen four times as often in affected children as in their unaffected siblings points to their likely contributory role in the illness.
Another striking asymmetry: when spontaneous CNVs contributing to autism were seen in girls, they tended to be much larger and hit many more genes—15 genes per CNV on average for girls vs. only 2 for boys. This touches on three observed phenomena: the preponderance of boys who become symptomatic; the preponderance of boys who have ASD but are “high-functioning”; and the comparative severity of the illness when it does occur in girls.
Since girls and boys are equally susceptible to DNA damage and new mutation—the rate of new mutation that damages a gene in humans is about one gene per three births—they should each have about the same number of CNVs. Presuming this is the case, why do more boys than girls develop ASD? And why are the observed CNVs in girls larger?
Wigler hypothesizes that females are in some manner “resistant” to autism, and that it takes a much larger, more devastating genomic hit to induce pathology in girls as compared with boys. The cause of this apparent female resistance to comparatively small genetic hits remains one of autism’s mysteries.
It has been speculated that unequal rates of brain development in the sexes may account for the smaller number of females with ASD. Wigler’s team discusses this possibility, noting that females “have an accelerated timescale for a number of cognitive milestones, for example, their first words at an earlier age. A quicker pace of development might reflect a robustness that offers females protection.”
Another mystery concerning gender draws attention to the status of adult women of child-bearing age who might be considered “carriers.” These would be women whose genomes contain transmissible mutations that can cause autism, yet who do not themselves manifest symptoms of ASD. The team speculates that the “high-risk” autism genes they carry may generate other effects in these women. They “may encounter difficulties at later stages of their lives that manifest as a different diagnostic category, or one that reduces fecundity.” If this is true, the disorder is “most likely to be one with a gender bias opposite that of ASD,” such as anorexia nervosa, Wigler proposes.
The team’s next insights about autism genetics will be made possible by a major technological advance—a shift from microarray detection of CNVs to the sequencing of the gene-encoding portions of the genome, or “exome,” of families in the Simons Simplex Collection (which will expand to include 3000 families). This work and work at other institutions of a similar nature should “crack open the field of autism genetics in two years,” says Wigler, whose own team has been gearing up for the sequencing phase during the last year.
Written by: Peter Tarr, Senior Science Writer | publicaffairs@cshl.edu | 516-367-8455
Citation
“Rare de novo and transmitted copy number variation in autistic spectrum disorders” will be published June 9 in Neuron. The authors are: Dan Levy, Michael Ronemus, Boris Yamron, Yoon-ha Lee, Anthony Leotta, Jude Kendall, Steven Marks, B. Lakshmi, Deepa Pai, Kenny Ye, Andreas Buja, Abba Krieger, Seungtai Yoon, Jennifer Troge, Linda Rodgers, Ivan Iossifov and Michael Wigler.
”Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses” will be published June 9 in Neuron. The authors are: Sarah R. Gilman, Ivan Iossifov, Dan Levy, Michael Ronemus, Michael Wigler and Dennis Vitkup.
Both papers are available online at: http://www.cell.com/neuron
Principal Investigator

Michael Wigler
Professor
Russell and Janet Doubleday Professor of Cancer Research
Cancer Center Member
Ph.D., Columbia University, 1978