Study suggests a unified model for how DNA is read, offering insight into how genes evolve
Cold Spring Harbor, NY — There are roughly 20,000 genes and thousands of other regulatory “elements” stored within the three billion letters of the human genome. Genes encode information that is used to create proteins, while other genomic elements help regulate the activation of genes, among other tasks. Somehow all of this coded information within our DNA needs to be read by complex molecular machinery and transcribed into messages that can be used by our cells.
Usually, reading a gene is thought to be a lot like reading a sentence. The reading machinery is guided to the start of the gene by various sequences in the DNA—the equivalent of a capital letter—and proceeds from left to right, DNA letter by DNA letter, until it reaches a sequence that forms a punctuation mark at the end. The capital letter and punctuation marks that tell the cell where, when, and how to read a gene are known as regulatory elements.
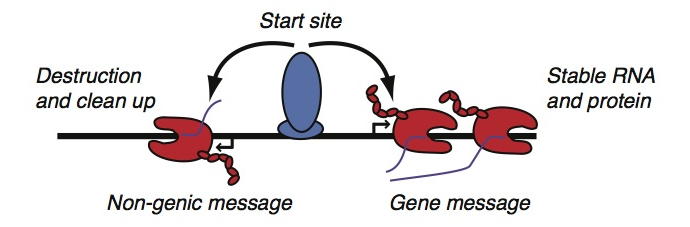
But scientists have recently discovered that genes aren’t the only messages read by the cell. In fact, many regulatory elements themselves are also read and transcribed into messages, the equivalent of pronouncing the words “capital letter,” “comma,” or “period.” Even more surprising, genes are read bi-directionally from so-called “start sites”—in effect, generating messages in both forward and backward directions.
With all these messages, how does the cell know which one encodes the information needed to make a protein? Is there something different about the reading process at genes and regulatory elements that helps avoid confusion? New research, published today in Nature Genetics, has revealed that the initial steps of the reading process itself are actually remarkably similar at both genes and regulatory elements. The main differences seem to occur after this initial step, in the length and stability of the messages. Gene messages are long and stable enough to ensure that genes becomes proteins, whereas regulatory messages are short and unstable, and are rapidly “cleaned up” by the cell.
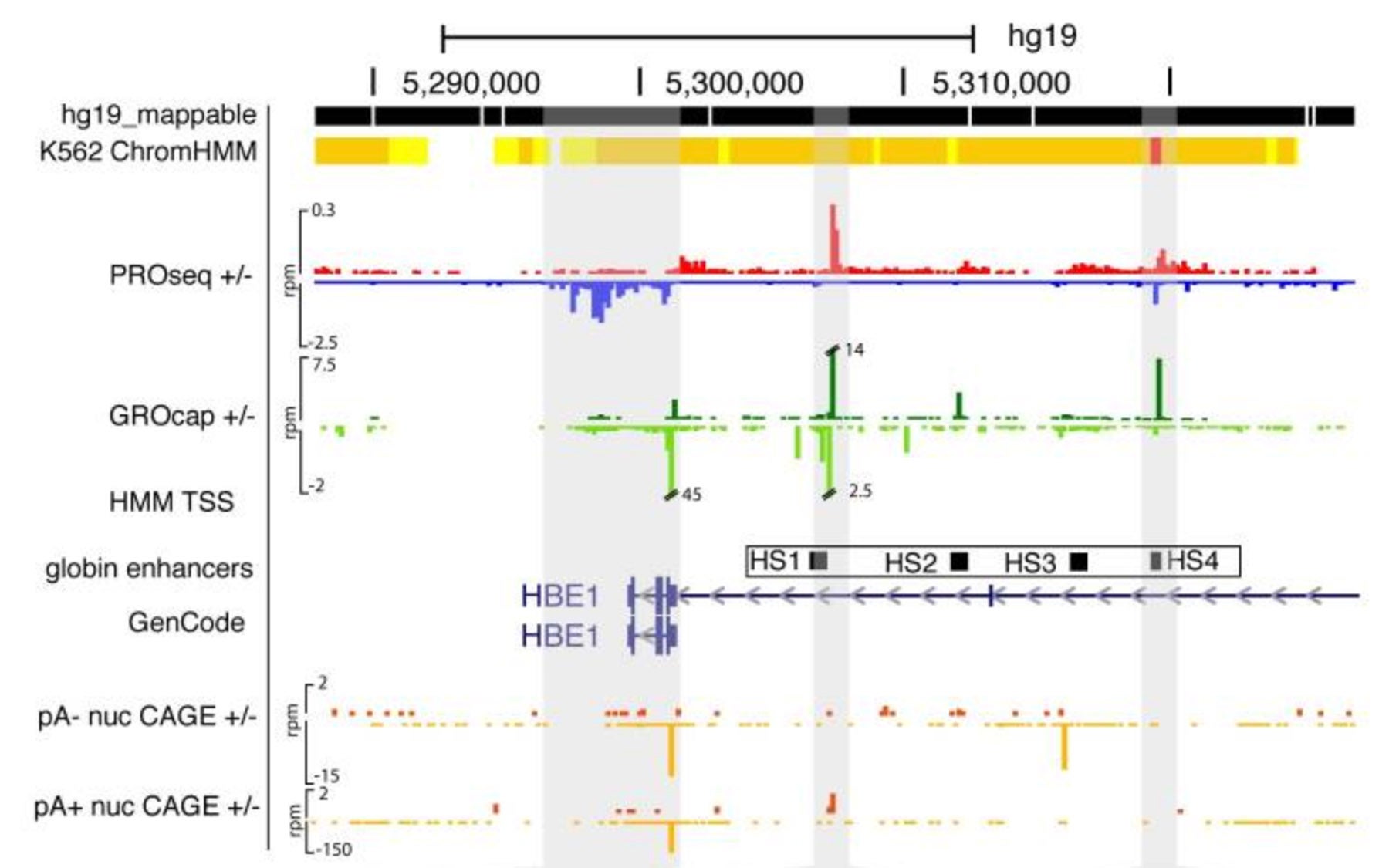
To make the distinction, the team, which was co-led by CSHL Professor Adam Siepel and Cornell University Professor John Lis, looked for differences between the initial reading processes at genes and a set of regulatory elements called enhancers. “We took advantage of highly sensitive experimental techniques developed in the Lis lab to measure newly made messages in the cell,” says Siepel. “It’s like having a new, more powerful microscope for observing the process of transcription as it occurs in living cells.”
Remarkably, the team found that the reading patterns for enhancer and gene messages are highly similar in many respects, sharing a common architecture. “Our data suggests that the same basic reading process is happening at genes and these non-genic regulatory elements,” explains Siepel. “This points to a unified model for how DNA transcription is initiated throughout the genome.”
Working together, the biochemists from Lis’s laboratory and the computer jockeys from Siepel’s group carefully compared the patterns at enhancers and genes, combining their own data with vast public data sets from the NIH’s Encyclopedia of DNA Elements (ENCODE) project. “By many different measures, we found that the patterns of transcription initiation are essentially the same at enhancers and genes,” says Siepel. “Most RNA messages are rapidly targeted for destruction, but the messages at genes that are read in the right direction—those destined to be a protein—are spared from destruction.” The team was able to devise a model to mathematically explain the difference between stable and unstable transcripts, offering insight into what defines a gene. According to Siepel, “Our analysis shows that the ‘code’ for stability is, in large part, written in the DNA, at enhancers and genes alike.”
This work has important implications for the evolutionary origins of new genes, according to Siepel. “Because DNA is read in both directions from any start site, every one of these sites has the potential to generate two protein-coding genes with just a few subtle changes. The genome is full of potential new genes.”
Written by: Jaclyn Jansen, Science Writer | publicaffairs@cshl.edu | 516-367-8455
Funding
This work was supported by the National Institutes of Health.
Citation
“Analysis of transcription start sites from nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers.” appears online in Nature Genetics on November 10, 2014. The authors are: Leighton Core, André Martins, Charles Danko, Colin Waters, Adam Siepel, and John Lis. The paper can be obtained online at: http://dx.doi.org/10.1038/ng.3142
Principal Investigator

Adam Siepel
Professor
Cancer Center Member
Ph.D., University of California, Santa Cruz, 2005