Aggressive tumors lacking p53 protein stop dead in their tracks when p53’s sister protein—TAp63—steps in
Cold Spring Harbor, NY — Oncologists have had their hands tied because more than half of all human cancers have mutations that disable a protein called p53. As a critical anti-cancer watchdog, p53 masterminds several cancer-fighting operations within cells. When cells lose p53, tumors grow aggressively and often cannot be treated.
These tumors might be tough, but they’re not invincible, suggests a new study from Cold Spring Harbor Laboratory (CSHL). The chink in the tumors’ armor, according to CSHL Associate Professor Alea Mills, Ph.D., is a protein called TAp63, an older sibling of p53 that’s usually intact and not mutated in most cancers.
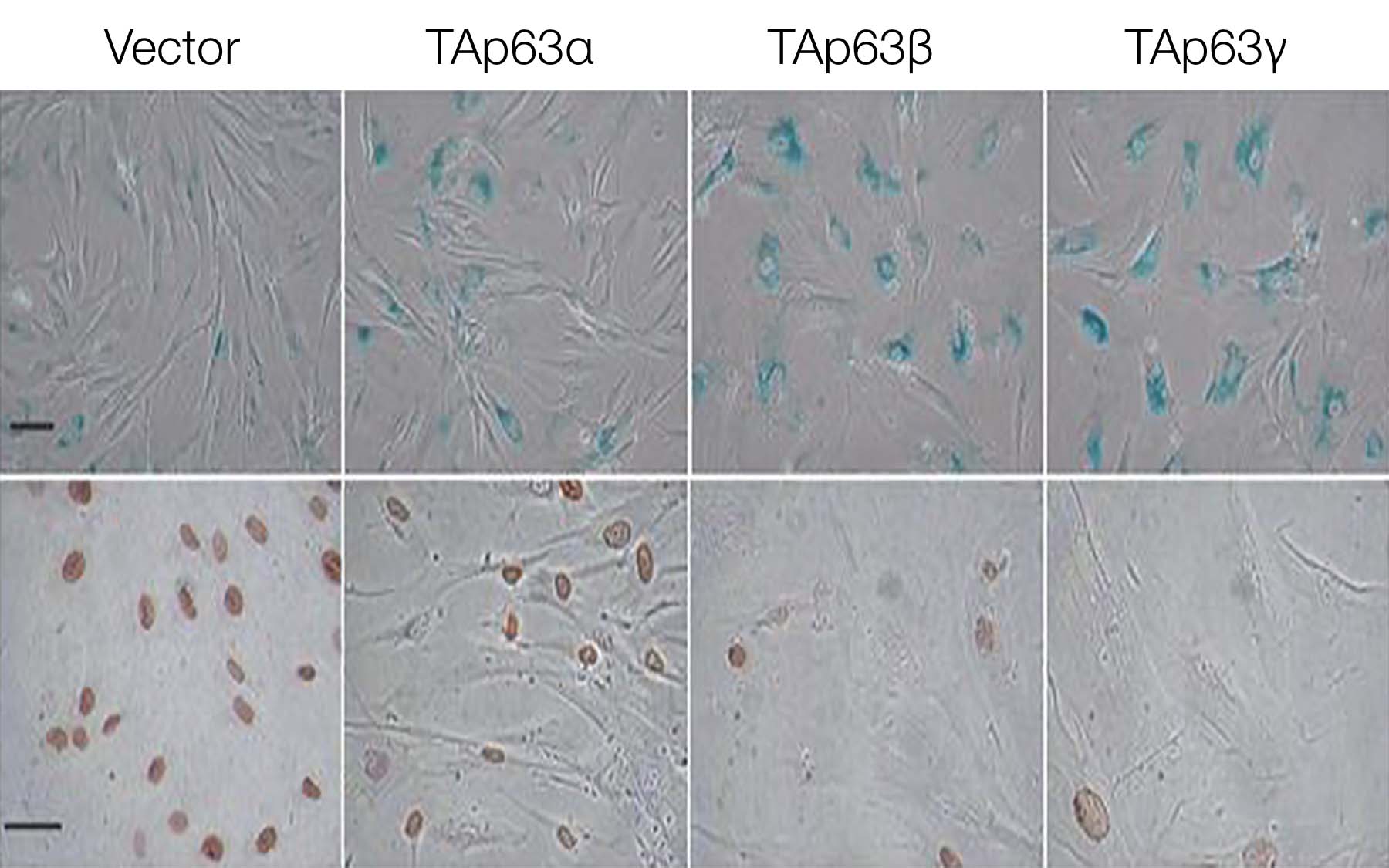
Mills and her team have succeeded in shutting off the growth of tumors in which p53 is missing by turning up the production of TAp63 proteins, which make up one class of proteins produced by the p63 gene. TAp63 completely blocked tumor initiation, the team found, by inducing senescence, a state of growth arrest in which tumor cells are still metabolically alive but fail to divide. More importantly, turning up the levels of TAp63 in cells that did not have p53 blocked the progression of established tumors in mice.
“We were very excited to see that TAp63 shuts down cancer completely independently of p53,” says Mills. “This means that we now have a way of attacking cancers that have damaged p53, which are very difficult to treat in the clinic.” The study, funded by a Research Scholar Award from the American Cancer Society, appears online ahead of print on November 8th in Nature Cell Biology.
TAp63 protects from cancer
“p63 is a double-edged sword,” says Mills, who discovered it when she was a postdoctoral researcher almost a decade ago. Of the six different proteins that are produced from the p63 gene, three promote activities that could lead to cancer. The remaining three, which are TAp63 versions, do the opposite: prevent cancer by triggering senescence—a process that shuts down the tumor.
Using a genetic maneuver called chromosome engineering, the CSHL team has found that TAp63 staves off cancer via senescence. This maneuver enabled them to specifically wipe out the TAp63 proteins while leaving the other p63 proteins intact. When exposed to the cancer-causing protein Ras, normal cells underwent senescence and could not form tumors. But cells without TAp63 failed to undergo senescence and developed into massive tumors.
When p53 and TAp63 were both missing, Ras caused extremely rapid and aggressive tumors. This tumor growth was much more severe than in tumors lacking either TAp63 or p53, suggesting that the two related proteins, working together, pack a stronger anti-cancer punch than either one alone.
A future anti-cancer strategy?
The team found that TAp63 could also substitute for p53 in its ability to halt tumor growth. When TAp63 was turned on in Ras-producing cells that were missing p53, tumors never started. “This suggests that TAp63 overrides cancer-promoting signals and prevents cancer from even forming,” explains Mills.
The team went a step further and showed that in addition to blocking the initiation of tumors, TAp63 could also shut down tumors that were already established. With help from Professor Scott Lowe, Ph.D., another researcher at CSHL, Mills’ team genetically tricked tumors into producing TAp63 when they were exposed to a compound called doxycycline.
Now being able to turn on TAp63 at will, the team transplanted these cells (which lack p53 and overproduce Ras) into mice and monitored for tumors. Once the tumors formed, a “mouse clinic” was set up: half of the “patients” were treated with doxycycline to induce TAp63 production, whereas the other half received a placebo. Tumor growth continued in the placebo group, with the tumors becoming five times larger within a week. In contrast, the tumors in the mice exposed to doxycycline were abruptly shut down, and the tumors even shrank in size. Mills speculates that the tumor cells disappear because the newly senescent cells might attract the attention of the immune system, which have the ability to destroy them.
Mills proposes that robustly activating TAp63 might be a viable anti-cancer strategy in the future. Alternatively, finding ways to stabilize the TAp63 that is already being made in cells or blocking pathways that combat TAp63 activities might also work, she speculates.
Written by: Communications Department | publicaffairs@cshl.edu | 516-367-8455
Citation
“TAp63 induces senescence and suppresses tumorigenesis in vivo,” was published online ahead of print on November 8th in Nature Cell Biology. The full citation is: Xuecui Guo, William M. Keyes, Cristian Papazoglu, Johannes Zuber, Wangzhi Li, Scott W. Lowe, Hannes Vogel and Alea A. Mills. The paper can be viewed at http://www.nature.com/ncb/journal/vaop/ncurrent/full/ncb1988.html (doi:10.1038/ncb1988).
Principal Investigator

Alea A. Mills
Professor
Cancer Center Member
Ph.D., University of California, Irvine, 1997