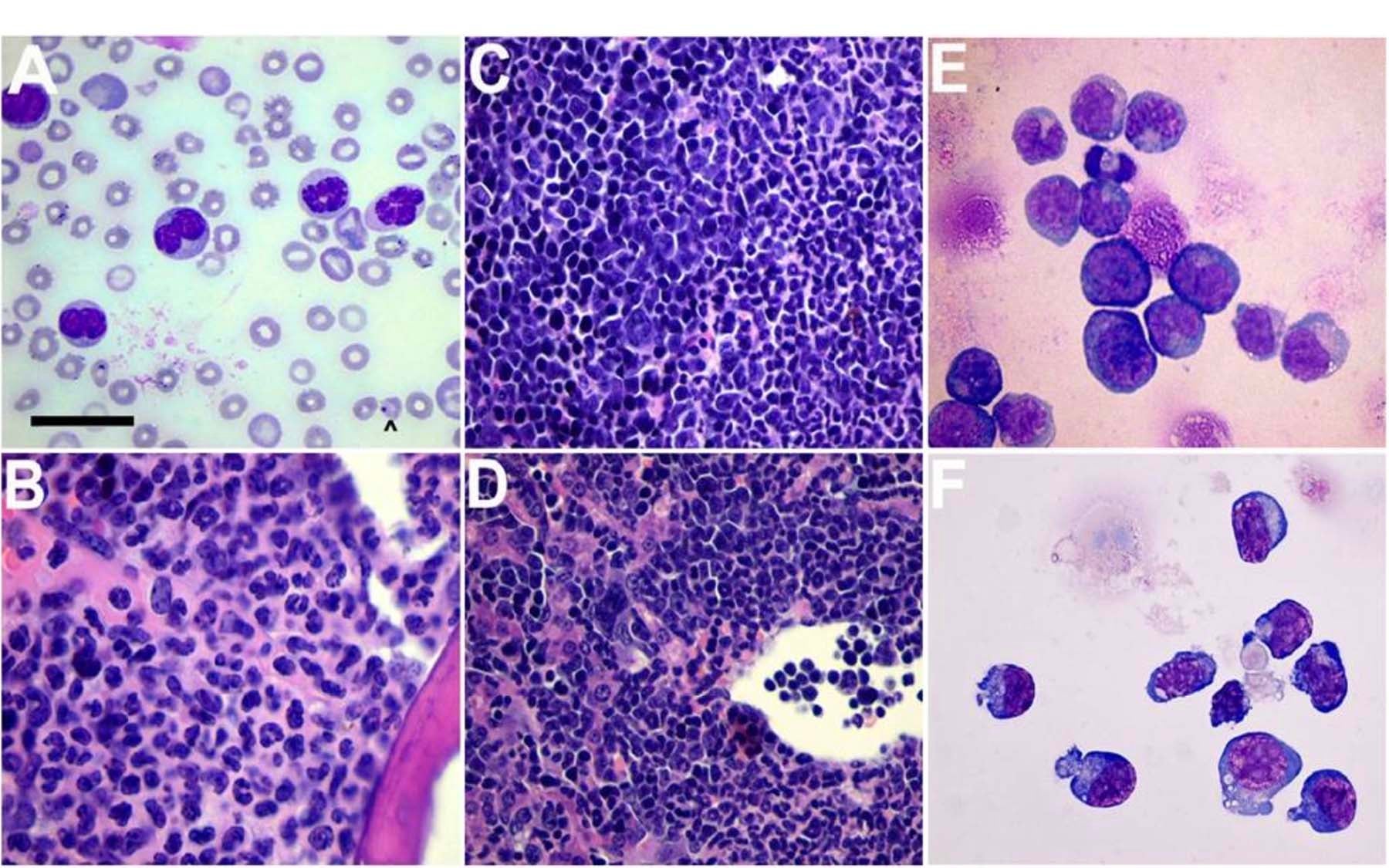
p53 loss promotes acute myeloid leukemia (AML) by enabling aberrant self-renewal of myeloid precursors
Cold Spring Harbor, NY — New research by scientists at Cold Spring Harbor Laboratory (CSHL) has illuminated in fine detail one of the genetic paths that leads to a particularly aggressive form of leukemia.
CSHL Professor Scott W. Lowe, Ph.D., an Investigator of the Howard Hughes Medical Institute, led a team of scientists who wanted to know more about how the absence of an important tumor-suppressing protein called p53 combines with another genetic “hit” in immature blood cells to give rise to acute myeloid leukemia, or AML. In experiments in living mice, the team discovered that if p53 is disabled in immature blood-cell “precursors” in which a mutation in a gene called Kras is also present, a built-in braking mechanism fails to engage and the cells proliferate out of control.
Mutations in p53, the gene that encodes the p53 “master tumor-suppressor” protein, had previously been associated with drug resistance and adverse outcome in AML. The mechanism, however, was a matter of conjecture prior to the new results, which are published in the July 1 issue of Genes & Development.
“Our team has shown how mutations in Kras and p53 act to reinforce one another to change the character of blood precursor cells, transforming them into cells that can renew themselves—and thus proliferate—indefinitely, somewhat as cancer stem cells are theorized to do,” says Lowe.
Graduate student Zhen Zhao, postdoctoral researcher Johannes Zuber, M.D., along with Lowe and others, hypothesized that loss of the p53 protein could impair the programmed cell fate and mortality of myeloid progenitors. “Although myeloid progenitors and their differentiated progeny normally lack self-renewal capabilities,” explains Zhao, “these cells apparently acquire this ability during the course of leukemia development. How this happens is what we set out to discover.”
Zhao and colleagues knew that mutations in Kras and other genes in the Ras signaling pathway can trigger replication stress in immature blood cells, in some instances calling p53 into action. That protein possesses a range of biological activities that are thought to contribute to its role in tumor suppression, including the ability to induce a cell to commit suicide (apoptosis) or to enter a quiescent state in which it can no longer multiply (senescence). When the p53 protein is missing or inactive, for instance, due to a mutation in the gene that encodes it, a potentially vital element in a cell’s “braking” system is disabled. Indeed, “mutational disruption of the p53 network is thought to occur in virtually all aggressive end-stage cancers,” notes Zhao, although mutations are seen in only 10%-15% of AML cases at diagnosis. Those tend to be the cases most resistant to therapy and most likely to be lethal.
Using so-called mosaic mouse models, the Lowe lab recently demonstrated that p53 mutations could specifically confer resistance to chemotherapeutic agents used to treat AML in human patients. Zhao, Zuber and colleagues took advantage of these genetically hybrid mice and a technology called RNA interference, or RNAi. Using a short-hairpin RNA molecule, they inactivated the p53 gene in hematopoietic cells that also had mutations that made Kras oncogenic. Myeloid precursor cells with only the Kras mutation are proliferative, but not self-renewing. But the team observed that cells with oncogenic Kras combined with a p53 deficiency did acquire the capacity to indefinitely self-renew, and transform into leukemia-initiating cells.
“Mice with one or the other mutation do develop T-cell malignancies, albeit ones with long latencies,” Zhao says. “But we showed how suppression of p53 cooperates with oncogenic Kras to promote AML of the treatment-resistant type. Importantly, we found that loss of p53, independently of oncogenic Kras activation, promotes the proliferation of myeloid progenitors.” It is the combination of p53 loss and activation of oncogenic Kras that confers upon these cells the ability to continually self-renew—and thus give rise to aggressive AML.
“In this work, we discovered a new tumor-suppressing function of p53, distinct, for instance, from apoptosis, and somewhat related to senescence,” says Zuber. “We showed that it has the ability to reinforce cell-fate and differentiation programs. In AML, p53 loss leads to cancer by disabling this reinforcement. The question we are now eager to answer is whether p53 has similar functions in other types of cancer.”
Written by: Peter Tarr, Senior Science Writer | publicaffairs@cshl.edu | 516-367-8455
Citation
“p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal” appears in Genes & Development July 1. The full list of authors is: Zhen Zhao, Johannes Zuber, Ernesto Diaz-Flores, Laura Lintault, Scott C. Kogan, Kevin Shannon and Scott W. Lowe. The paper can be obtained online at: http://genesdev.cshlp.org