The next “next-gen” technology in genome sequencing has gotten a major boost.
Cold Spring Harbor, NY — A quantitative biologist at Cold Spring Harbor Laboratory (CSHL) and collaborators have published results of experiments that demonstrate the power of so-called single-molecule sequencing, which was recently introduced but whose use has so far been limited by technical issues.
The team, led by CSHL Assistant Professor Michael Schatz and Adam Phillippy and Sergey Koren of the National Biodefense Analysis and Countermeasures Center and the University of Maryland (UMD), has developed a software package that corrects a serious problem inherent in the newest sequencing technology: the fact that every fifth or sixth DNA “letter” it generates is incorrect. The high error rate is the flip side of the new method’s chief virtue: it generates much longer genome “reads” than other technologies currently used, up to 100 times longer, and thus can provide a much more complete picture of genome structure than can be obtained with current, “2nd-gen” sequencing technology.
Using mathematical algorithms, Schatz and the team have preserved the great advantage of the “3rd-gen” method while all but eliminating its chief flaw. They have reduced the error rate from about 15% or greater to less than one-tenth of one percent. This mathematical “fix”—which has been published in open-source code to the World Wide Web—greatly increases the practical utility of 3rd-gen sequencing for the entire biomedical research community.
The team demonstrates the breadth of potential applications of single-molecule sequencing by applying their fix to sequencing tasks ranging from the tiny bacteriophage virus at one end of the difficulty scale to the large and vastly more complex genome of the parrot, at the other. The parrot genome is more than a third the size of the human genome and is published online July 1 with the team’s paper in Nature Biotechnology. The parrot sequence is “far superior to that of any previously sequenced bird genome,” Schatz says.
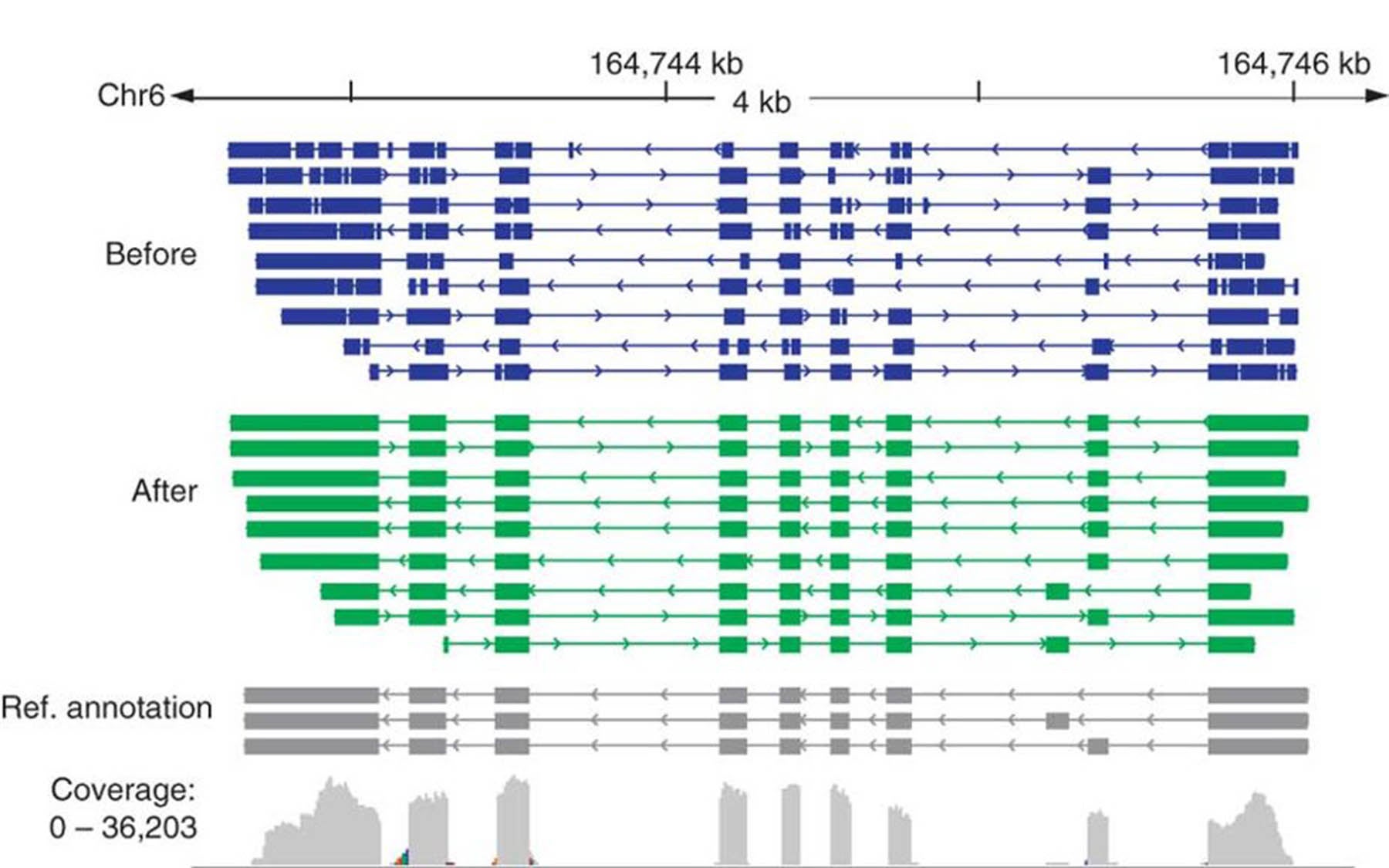
To understand why it is better is to appreciate the advantages of 3rd-gen sequencing. The main advantage has to do with the average length of each “read” (i.e., genome segments read by a sequencer). The individual sequences are assembled into “contigs”—shorthand for contiguous sequences—much the way pieces in a jigsaw puzzle are assembled. In currently used 2nd-gen technology, the contigs are very small, and are massively redundant. A “consensus” version of each segment, representing the results of many layered reads, tends to be extremely accurate. But the small size of puzzle pieces prevents accurate assembly of certain genome portions, like those containing long repetitive sequences.
Obtaining superior versions of complete genomes was the objective that motivated Schatz and his collaborators, who also include HHMI Investigator Erich D. Jarvis of Duke University and CSHL Professor W. Richard McCombie, a sequencing pioneer, among others.
Combining the best of both generations
With single-molecule sequencing, the assembled contigs are much longer—affording a much better picture of relatively larger genome segments, including those occupied by lengthy repeats. This is what Schatz and his team wanted to preserve, while at the same time boosting the error-free rate. They did so by effectively taking the best of both 2nd- and 3rd-gen technologies.
“We call our approach ‘hybrid error correction,’” Schatz explains.
The team’s major insight was to take advantage of the long-read data offered by a 3rd-gen machine like that used in their experiments, a Pacific Biosciences RS sequencer, and mixing in highly accurate short reads obtained from a separate 2nd-gen sequencer. The two data types were run through an open-source genome assembly program called Celera Assembler to generate a clean final assembly that has proven 99.9% error-free and composed of contigs whose median size is at least double that obtainable with 2nd-gen “short-read” sequencers. Contig sizes are expected to increase appreciably in subsequent iterations of the hybrid approach as single molecule long-read sequencing improves.
High-quality genome assemblies are especially important for genome annotation and comparative genome analyses. Many microbial genome analyses depend on finished genomes, but their cost is prohibitive using older technologies. A high-quality analysis of the genomes of higher organisms depends upon continuous sequences that capture long stretches of DNA that spell out genes. Discoveries in recent years of spontaneously occurring structural changes in genomes called copy number variations—such as those made by CSHL Professor Mike Wigler and his team in their research on schizophrenia and autism—make clear the importance of being able to obtain clean and accurate pictures of the entire genomes of affected individuals.
With hybrid error correction, Schatz and his colleagues have “demonstrated that high error rates associated with long reads need not be a barrier to genome assembly,” he summarizes. “High-error long reads can be efficiently assembled in combination with complementary short reads to produce assemblies not previously possible.”
Written by: Peter Tarr, Senior Science Writer | publicaffairs@cshl.edu | 516-367-8455
Funding
The following entities provided support for this research and those who performed it: U.S. Dept. of Homeland Security; U.S. Dept. of Energy; National Institutes of Health; National Science Foundation; Howard Hughes Medical Institute.
Citation
“Hybrid error correction and de novo assembly of single-molecule sequencing reads” appears online in Nature Biotechnology, July 1, 2012. The authors are: Sergey Koren, Michael C. Schatz, Brian P. Walenz, Jeffrey Martin, Jason Howard, Ganeshkumar Ganapathy, Zhong Wang, David A. Rasko, W. Richard McCombie, Erich D. Jarvis and Adam M. Phillippy. The paper can be obtained online at http://www.nature.com/nbt/journal/vaop/ncurrent/full/nbt.2280.html.
