Cold Spring Harbor, NY — The genome of the corn plant—or maize, as it’s called almost everywhere except the US—“is a lot more exciting” than scientists have previously believed. So says the lead scientist in a new effort to analyze and annotate the depth of the plant’s genetic resources.
“Our new research establishes the amazing diversity of maize, even beyond what we already knew was there,” says Doreen Ware, Ph.D., of the US Department of Agriculture and Cold Spring Harbor Laboratory (CSHL) in New York. “This diversity is fascinating in its own right and at the same time has great import for agriculture.” Maize is one of the world’s top-three staple foods; along with rice and wheat, it accounts for two-thirds of world food consumption.
Ware was part of a large multinational team that in 2009 assembled the first-ever sequence of maize’s 30,000 or so genes, based on a single variety called B73. The discovery of maize’s extraordinary protein diversity is based on more accurate “long-read” sequencing technology, provided through a research partnership with PacBio, a sequencing company. This updated technology did not reveal very many previously unknown genes, but rather, many more of the RNA messages that are generated when genes are expressed, i.e., activated.
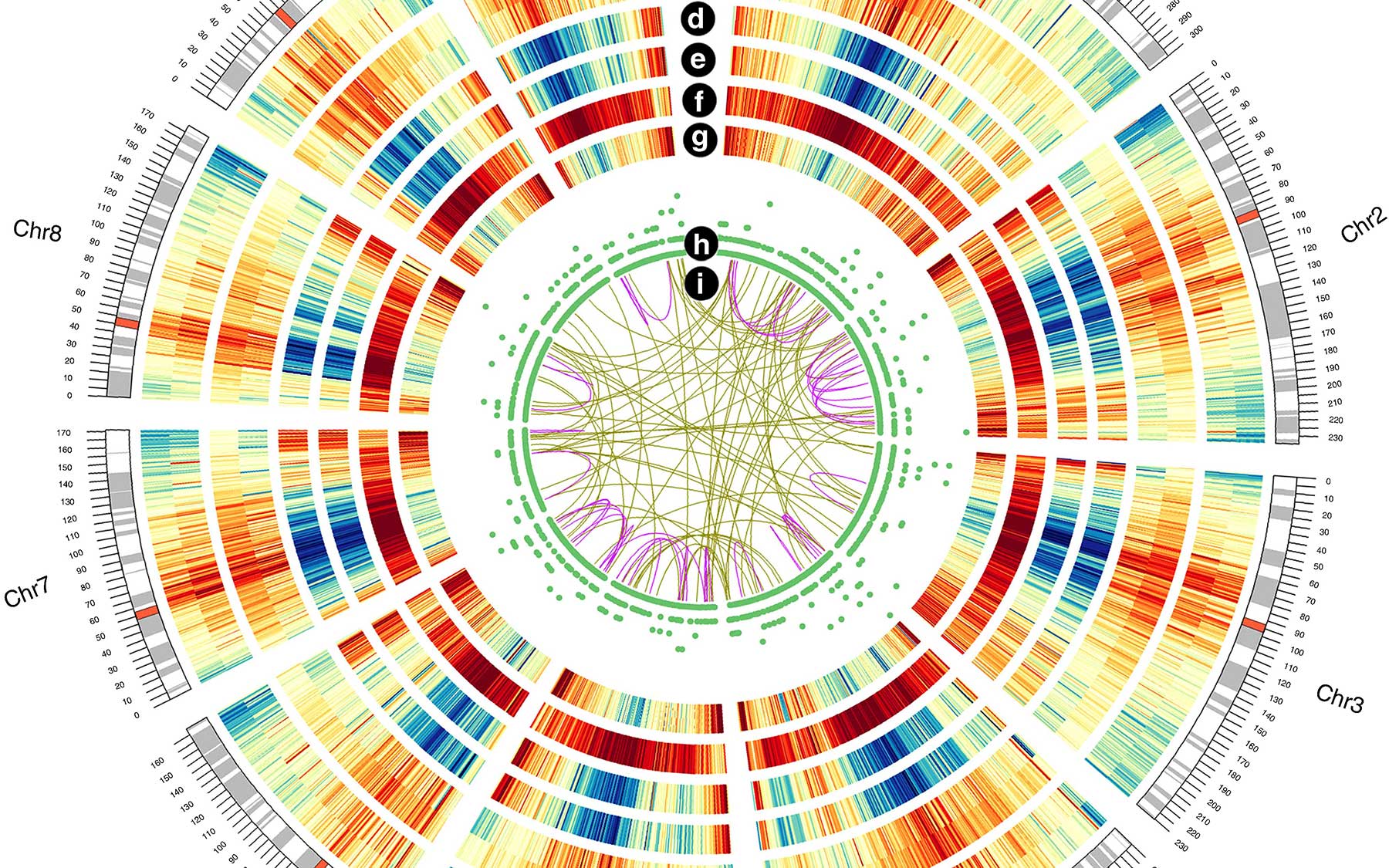
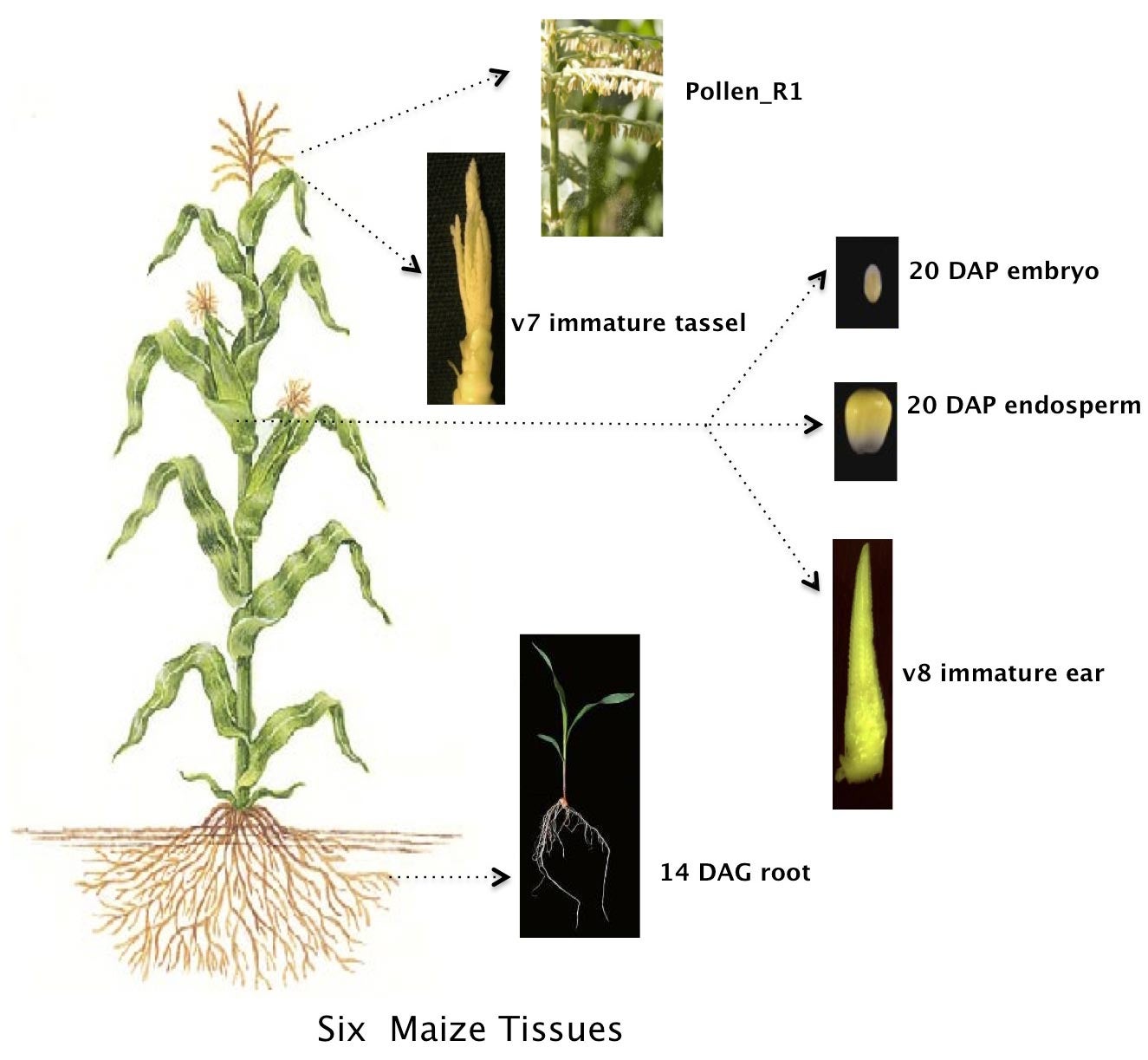
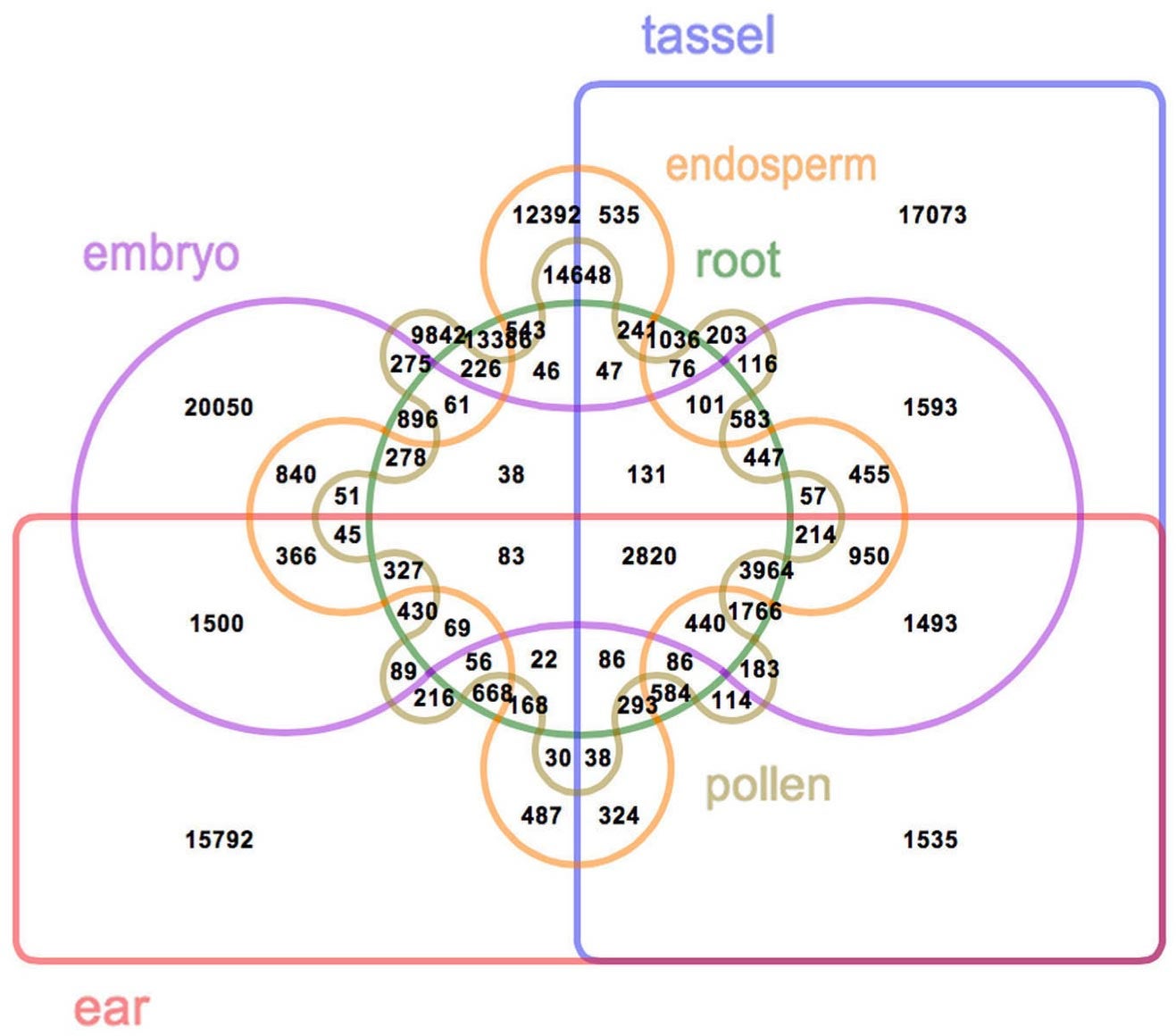
In all, 111,151 RNA transcripts from genes being expressed in six different maize tissues were read and analyzed in the research. About 57% of these messages had never been seen—and therefore had never been sequenced. “These were the messages that told us that our efforts to annotate and characterize the 2009 maize reference genome have been far from complete,” says Bo Wang, Ph.D., a postdoctoral investigator in Ware’s lab and first author of the paper reporting the new research.
What makes one maize plant potentially much different from any other, even individuals of the same variety, is the way its genes are capable of being expressed, depending on conditions both internal to the plant and in the surrounding environment, e.g., levels of soil moisture, nutrients, or available light.
Many of maize’s total complement of 30,000-odd genes can generate RNA messages that can be spliced, i.e., edited, in different ways, leading to the production of different proteins—proteins with different shapes and different functions. Alternative RNA splicing is a phenomenon that occurs in nearly all forms of multicellular life, including humans. The current work begins to suggest how extensively alternative splicing, and therefore alternative developmental possibilities, are available to each maize individual—a surprise, indeed, for anyone who thought the 2009 genome annotation captured the essence of the species.
The practical import of the research is that “it begins to reveal new functional parts that we didn’t know about before,” says Ware. “By having insight into what those other parts are and what they do, we begin to realize new ways of breeding corn, adapting it, for example, to changes in climate as average annual temperatures in growing zones continue to climb.”
Written by: Peter Tarr, Senior Science Writer | publicaffairs@cshl.edu | 516-367-8455
Funding
This research was funded by the National Science Foundation Improving Plant Genome Annotation grant #1127112; NSF Cereal Gene Discovery grant #1032105; US Department of Agriculture ARS CRIS 1907-21000-030-00D; and NSF Rare Alleles grant #1238014.
Citation
“Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing” appears online June 24, 2016 in Nature Communications. The authors are: Bo Wang, Elizabeth Tseng, Michael Regulski, Tyson A. Clark, Ting Hon, Yinping Jiao, Zhenyuan Lu, Andrew Olson, Joshua C. Stein and Doreen Ware. The paper can be viewed at: http://www.nature.com/ncomms/archive/category/article/index.html
