Scientists develop algorithm to uncover genomic insertions and deletions involved in autism, OCD

Cold Spring Harbor, NY — With three billion letters in the human genome, it seems hard to believe that adding a DNA base here or removing a DNA base there could have much of an effect on our health. In fact, such insertions and deletions can dramatically alter biological function, leading to diseases from autism to cancer. Still, it is has been difficult to detect these mutations. Now, a team of scientists at Cold Spring Harbor Laboratory (CSHL) has devised a new way to analyze genome sequences that pinpoints so-called insertion and deletion mutations (known as “indels”) in genomes of people with diseases such as autism, obsessive-compulsive disorder and Tourette syndrome.
The letters in the human genome carry instructions to make proteins, via a three-letter code. Each trio spells out a “word;” the words are then strung together in a sentence to build a specific protein. If a letter is accidentally inserted or deleted from our genome, the three-letter code shifts a notch, causing all of the subsequent words to be misspelled. These “frameshift” mutations cause the protein sentence to become unintelligible. Loss of a single protein can have devastating effects for cells, leading to dysfunction and sometimes to serious diseases.
DNA insertions and deletions vary in length and sequence. Each indel can range in size from one DNA letter to thousands, and they are often highly repetitive. Their variability has made it challenging to identify indels, despite major advancements in genome sequencing technology. They are, in effect, regions of the genome that have remained hidden from view as researchers search for the mutations that cause disease.
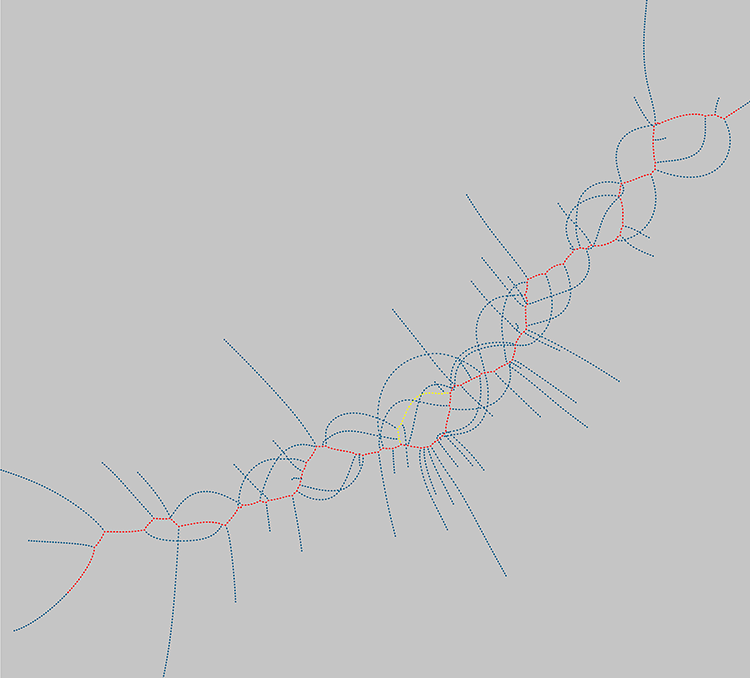
The inner workings of Scalpel. Each dot represents a short sequence of DNA from the reads mapped to a gene. The red path spells out the normal version of the gene, while the yellow path spells out a truncated path with the indel. The blue “spikes” show where there are other errors in the reads that have been mapped here. A team of CSHL scientists, including Assistant Professors Mike Schatz, Gholson Lyon, and Ivan Iossifov, and Professor Michael Wigler, has devised a way to mine existing genomic datasets for indel mutations. The method, which they call Scalpel, begins by grouping together all of the sequences from a given genomic region. Scalpel—a computer formula, or algorithm—then creates a new sequence alignment for that area, much like piecing together parts of a puzzle.
“These indels are like very fine cuts to the genome—places where DNA is inserted or deleted—and Scalpel provides us with a computational lens to zoom in and see precisely where the cuts occur,” says Schatz, a quantitative biologist. Such information is critical to understand the mutations that cause disease. In work published today in Nature Methods, the team used Scalpel to search for indels in patient samples. Lyon, a CSHL researcher who is also a practicing psychiatrist, worked with his team to analyze a patient with severe Tourette syndrome and obsessive-compulsive disorder, identifying and validating more than a thousand indels to demonstrate the accuracy of the method.
The CSHL team performed a similar analysis to search for indels that are associated with autism. They explored a dataset of 593 families from the Simons Simplex Collection, a group composed entirely of families with one affected child but no other family members with the disorder. While the researchers discovered a total of 3.3 million indels across the 593 families, most appeared to be relatively harmless. Still, a few dozen mutations stood out to be specifically associated with autism. “All this adds to our body of knowledge about the spontaneous mutations that cause autism,” says Schatz.
But the tool can be applied much more broadly. “We are collaborating with plant scientists, cancer biologists, and others, looking for indels,” says Schatz. “This is a powerful tool, and we are looking forward to revealing new pieces of the genome that make a difference, throughout the tree of life.”
Written by: Jaclyn Jansen, Science Writer | publicaffairs@cshl.edu | 516-367-8455
Funding
This work was supported by US National Institutes of Health, US National Science Foundation, the CSHL Cancer Center Support Grant, the Stanley Institute for Cognitive Genomics, and the Simons Foundation.
Citation
“Accurate de novo and transmitted indel detection in exome-capture data using microassembly” appears online in Nature Methods on August 17, 2014. The authors are: Giuseppe Narzisi, Jason O’Rawe, Ivan Iossifov, Han Fang, Yoon-ha Lee, Zihua Wang, Yiyang Wu, Gholson Lyon, Michael Wigler, and Michael Schatz. The paper can be obtained online at: http://dx.doi.org/10.1038/nmeth.3069
Principal Investigator

Michael Schatz
Adjunct Associate Professor