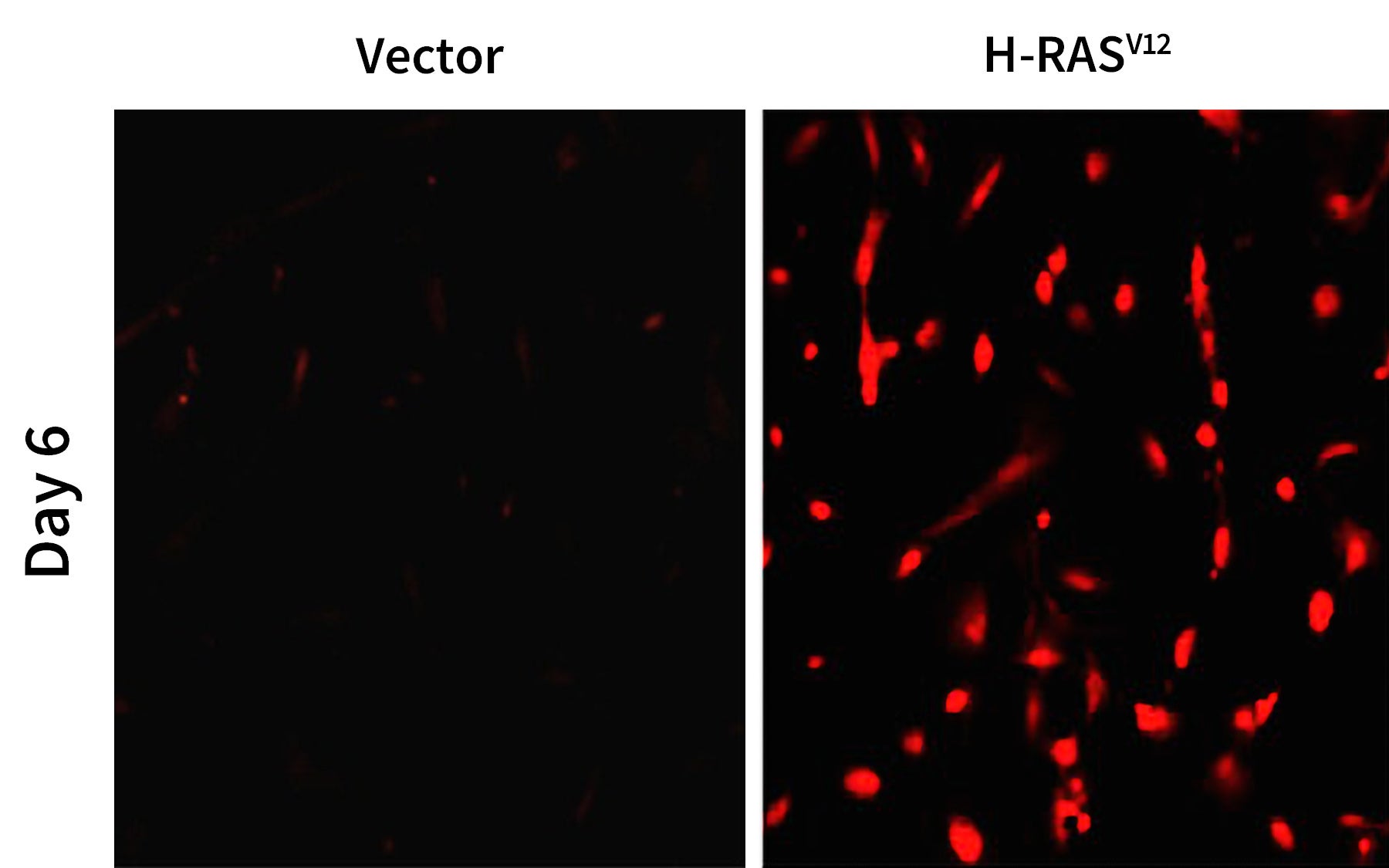
Cold Spring Harbor, NY — What happens inside cells when they detect the activation of a cancer-inducing gene? Sometimes, cells are able to signal internally to stop the cell cycle. Such cells are able to enter, at least for a time, a protective non-growth state.
Since the 1980s, scientists have known that mutations in a human gene called RAS are capable of setting cells on a path to cancer. Today, a team at Cold Spring Harbor Laboratory (CSHL) publishes experiments showing how cells can respond to an activated RAS gene by entering a quiescent state, called senescence.
CSHL Professor Nicholas Tonks and Benoit Boivin, now a University of Montreal Assistant Professor, co-led a team that traced the process in exquisite detail. They began by confirming that activation of mutant, oncogenic H-RAS, one of the human RAS oncogene variants, spurs cells to generate hydrogen peroxide (H2O2), a form of reactive oxygen species, or ROS. “Most people, when they think about ROS, think about the great damage they can do at high concentrations,” says Tonks. “But this research exemplifies how the controlled production of ROS in cells can play a beneficial role.”
The team showed how the production of ROS in response to oncogenic H-RAS enables cells to fine-tune signaling pathways, leading them to enter a senescent state. A key part of this process is the impact of ROS on a protein called PTP1B. Tonks discovered PTP1B some 25 years ago. It is an enzyme—one in a family of protein tyrosine phosphatases (PTPs), of which there are 105 in humans—that performs the essential biochemical task of removing phosphate groups from amino acids called tyrosines in other proteins. Adding and removing phosphates is one of the principal means by which signals are sent among proteins.
In cells with oncogenic H-RAS, ROS is produced in small quantities, sufficient to render PTP1B inactive. The team found that with the phosphate-removing enzyme unable to do its usual job, a key protein called AGO2 remains phosphorylated—with the consequence that it can no longer do what it normally does, which is engage the cell’s RNA interference machinery. In normal cells, the RNAi machinery represses a gene called p21. But in this specific condition —with H-RAS oncogenically activated, PTP1B inactivated by ROS, and RNAi disabled—p21 proteins begin to accumulate unnaturally, the team discovered.
“This is the key step—accumulation of p21 proteins effectively halts the cell cycle and enables the cell to enter the senescent state,” explains Ming Yang, a doctoral student in the Tonks lab. She and Astrid Haase, Ph.D., a postdoctoral investigator in the laboratory of CSHL Professor Greg Hannon, are the first two authors, respectively, on the team’s paper, published in Molecular Cell.
“This is confirmation of a hypothesis we presented five years ago,” Tonks says. “We knew that oncogenic RAS induced the production of ROS. We proposed that this would lead to regulation of PTPs, and using the example of PTP1B this is precisely what the team discovered in this work —showing also how inactivation of this PTP is part of a complex signaling cascade that can culminate in the induction of senescence.”
ROS have been linked to the pathogenesis of several diseases including Alzheimer’s, diabetes and heart failure. “By showing that PTP1B inactivation by oxidation prevents AGO2 from doing its job, we make a clear link between ROS and gene silencing which could also be observed in other pathologies” says Boivin. Hence, the role of PTP1B in keeping the RNAi machinery active could have important ramifications.
Entering senescence is not enough to arrest oncogenesis completely. Oncogenic mutations typically multiply as cancers evolve to promote their survival and proliferation. But the current work does show the potential importance of knowing the genetic background of a cancer patient, for there are windows of time—narrow though they may be—in which naturally occurring processes induce pauses in growth.
Written by: Peter Tarr, Senior Science Writer | publicaffairs@cshl.edu | 516-367-8455
Funding
Funders of the research described here include: National Institutes of Health grants CA53840 and GM55989, and S10RR027990 and P30CA016087; CSHL Cancer Center Support Grant; Joni Gladowsky Breast Cancer Foundation; the Don Monti Memorial Research Foundation; The Irving A. Hansen Memorial Foundation; West Islip Breast Cancer Coalition for Long Island; Glen Cove CARES; Find A Cure Today (FACT); Constance Silveri; Robertson Research Fund; and Masthead Cove Yacht Club Carol Marcincuk Fund; Fonds de Recherche de l’Institut de Cardiologie de Montréal; Heart and Stroke Foundation-Québec.
Additional funders of research in the Tonks Lab at CSHL include: Aboff’s Inc.; Hearts for Cancer; Islip Breast Cancer Coalition; Jo-Ellen and Ira Hazan; Panera Bread; Fannie E. Rippel Foundation; Judi Shesh Memorial Foundation; Mary F. Smith Foundation; Caroline Bassett; The WTFC Organization, Inc.
Citation
“Dephosphorylation of Tyrosine 393 in Argonaute 2 by Protein Tyrosine Phosphatase 1B Regulates Gene Silencing in Oncogenic RAS-Induced Senescence” appears online in Molecular Cell August 28, 2014. The authors are: Ming Yang, Astrid D. Haase, Fang-Ke Huang, Gerald Coulis, Keith D. Rivera, Bryan C. Dickinson, Christopher J. Chang, Darryl J. Pappin, Thomas A. Neubert, Gregory J. Hannon, Benoit Boivin and Nicholas K. Tonks. The paper can be obtained at: http://www.cell.com/molecular-cell/newarticles
Principal Investigator

Nicholas Tonks
Professor
Caryl Boies Professor of Cancer Research
Cancer Center Associate Director of Shared Resources
Ph.D., University of Dundee, 1985