Cold Spring Harbor, NY — DNA has often been called “the book of life,” but this popular phrase makes some biologists squirm a bit. True, DNA bears our genes, which spell out the instructions our cells use to make proteins—those workhorse molecules that make just about everything in life possible.
But the precise relationship between the protein “blueprints” encoded in genes and the amount of protein a given cell actually makes is by no means clear. When a gene is activated and its message is copied into a molecule of RNA, a biologist can be no more certain of knowing if it results in the manufacture of a working protein than a banker is of knowing whether a check written by one of its customers will end up being cashed.
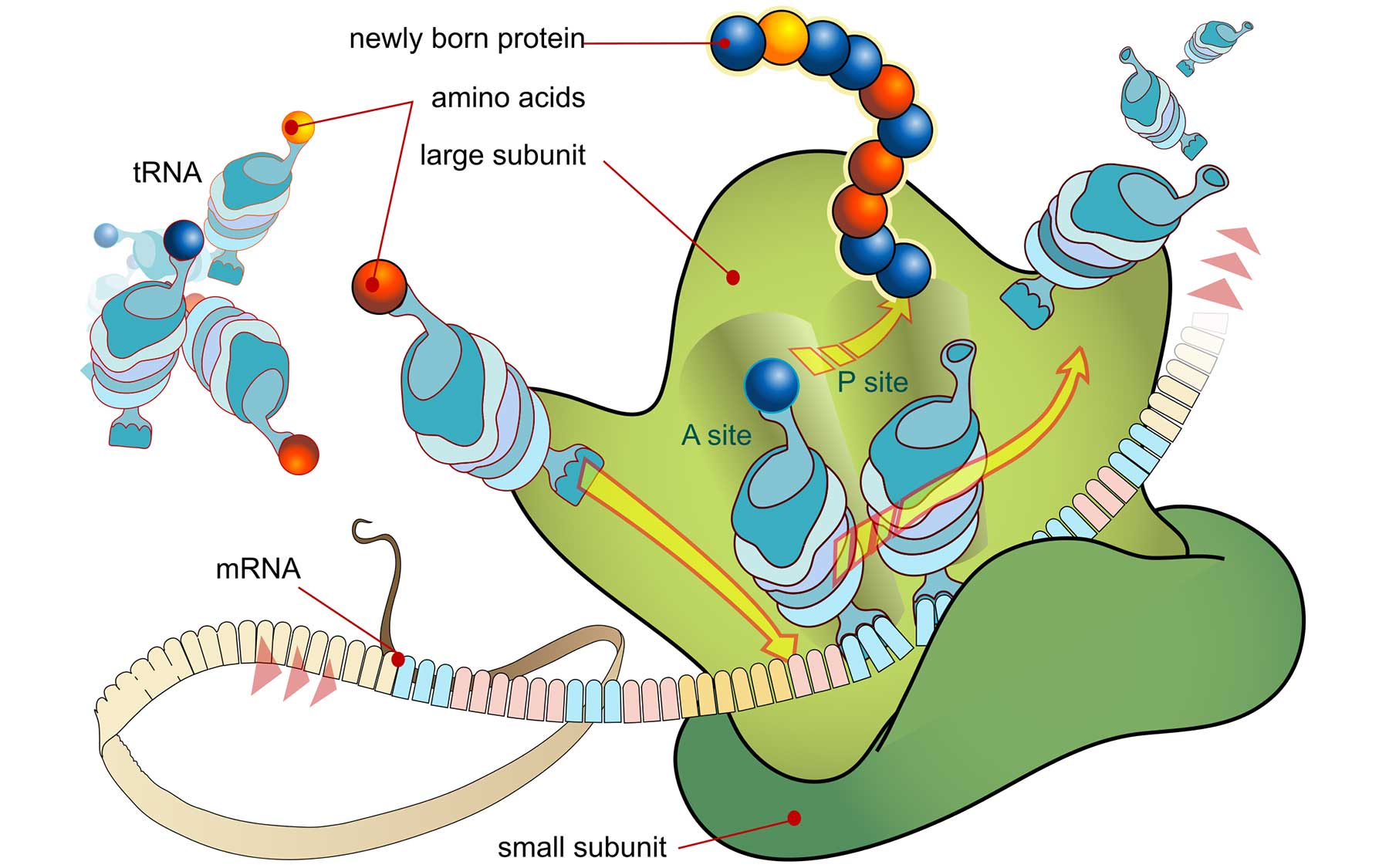
Thanks to advancements in DNA and RNA sequencing, biologists are incredibly good at knowing how much of a gene’s code is at any moment being copied into RNA messages, the first step in making protein. But they’re not so good at figuring out how quickly those RNA messages are actually read from end to end at cellular factories called ribosomes, where proteins are synthesized.
Now, a multidisciplinary team of researchers from Cold Spring Harbor Laboratory (CSHL), Stony Brook University (SBU) and Johns Hopkins University (JHU) has released software that can help biologists more accurately determine this. They used single-celled yeast and the common microbe E. coli to demonstrate their new program, called Scikit-Ribo.
Scikit-Ribo is like a set of mathematical corrective lenses designed to be “placed over” a method introduced in 2009, called Riboseq. The latter revealed as never before which, and how quickly, cells translate RNA into protein. It was a great advance, says Michael Schatz, Ph.D., a quantitative biologist at CSHL and JHU, who with Gholson Lyon, M.D., Ph.D., of CSHL, supervised the work of a talented young scientist Han Fang, Ph.D., a recent graduate of SBU. It was Fang who figured out how to construct the corrective lens so that the Riboseq data could be brought into focus.
Fang’s insight was to use advanced statistical modeling techniques to account for the fact that ribosomes do not work at a uniform rate, but rather tend to pause—for instance, when they encounter hairpin-shaped kinks in incoming RNA messages. Scikit-ribo also filters out noise that muddied raw Riboseq results. Now the two methods can be used together, to generate a much more accurate picture of which RNA messages are being read at specific ribosomes, and, perhaps most important, how much functional protein is being generated.
“The amount of protein that’s actually made may or may not be the same as the amount that a given gene is being expressed,” says Schatz. Having a more reliable way of knowing will help in disease research. CSHL’s Lyon used Scikit-Ribo to explore the ability of ribosomes to convert certain RNA messages into protein, in the context of a rare human developmental illness he discovered in 2011 called Ogden Syndrome. In this case, the new method was used to study the hypothesis that errors in translation at the ribosome may be involved in disease causation.
Written by: Peter Tarr, Senior Science Writer | publicaffairs@cshl.edu | 516-367-8455
Funding
NIH, NSF; CSHL Cancer Center Support Grant; NIGMS.
Citation
Fang H et al, “Scikit-ribo enables accurate estimation and robust modeling of translation dynamics at codon resolution,” appeared in Cell Systems on January 17, 2018.
