Team explains a key mechanism to prevent aberrant cell proliferation
Cold Spring Harbor, NY — It is one of the wonders of nature: multiplying cells are able to precisely copy their genetic material, once and only once, and spatially segregate the resulting two sets of chromosomes when the time comes to separate into two “daughter” cells. Just in our blood system alone, we have about 500 million cells born in the bone marrow every minute of our lifetime. Inside each of these cells, the DNA in chromosomes has to be accurately duplicated and then evenly segregated into the daughter cells as they divide.
In humans, the process is astonishingly complex—multi-layered, with checkpoints and redundancies built in, the work of eons of evolution minimizing the chance that something will go awry. And yet, we know very well that things do go wrong from time to time; errors in DNA replication and chromosome segregation are an important cause of cancer and other diseases.
The opposing effects of ORC1 and CDC6 in controlling levels of Cyclin E contributes to the stability of the genome. It is a mechanism for directly linking the process of DNA replication with a cell’s commitment to divide.
— CSHL Professor Bruce Stillman
New research from the laboratory of Professor Bruce Stillman, Ph.D., president of Cold Spring Harbor Laboratory (CSHL), sheds light on a critical decision that every newly born cell makes: whether to continue to proliferate or to exit the cell-division cycle. In cancer, the equivalent of an “on” switch is stuck in that position. The cell prepares immediately to divide again, bypassing checkpoints.
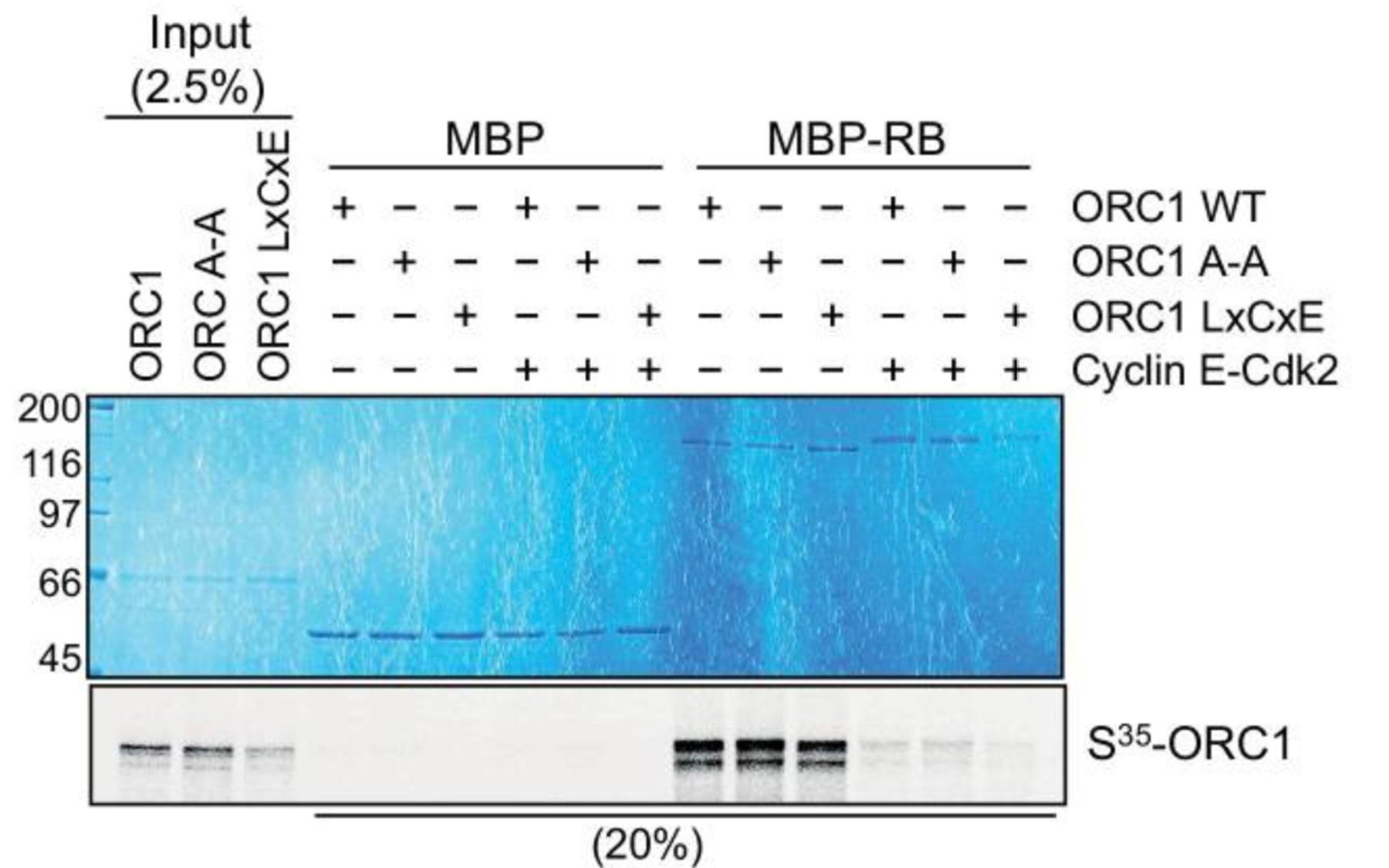
The decision to stop proliferating depends on delaying the expression of a protein called Cyclin E. Cyclin E and its partner, protein kinase CDK2, are key regulators of the decision of whether a cell will commit to a new round of cell division or remain in a non-proliferative state. Stillman and postdoctoral investigator Manzar Hossain, Ph.D., today publish in eLife the results of experiments demonstrating precisely how Cyclin E expression is kept in balance in normal cells by the opposing actions of two proteins.
The two proteins are called ORC1 and CDC6. As Stillman’s lab showed last year, during mitosis, when two daughter cells separate, each inherits chromosomes to which ORC1 is bound. Thus it is inherited into the new cells and can immediately act to control Cyclin E levels. Previously, the lab had shown that when ORC1 is missing, Cyclin E levels rise.
Now, Stillman and Hossain have discovered that ORC1 represses the activation of the gene (called CCNE1) that encodes instructions for manufacturing Cyclin E. This means that as the next cell cycle begins—a phase biologists call G1—Cyclin E initially is not expressed because ORC1 blocks the CCNE1 gene. The new cell thus has a period of time during which it can integrate signals that indicate that the time and conditions are right to enter into another round of cell division or decide not to divide again.
ORC1 is one of the main protein components of the machine called the Origin Recognition Complex, or ORC, which determines where along the chromosomes the process of duplicating the DNA begins. In 1992, Stillman and colleagues identified ORC for the first time, in yeast. Single-celled yeast are vastly less complex than human cells, having only a few hundred replication start sites in their DNA, but human cells have many tens of thousands of positions along their genetic material where ORC assembles the first proteins that are destined to copy DNA, called pre-RC complexes.
The new research reveals how ORC1’s anti-proliferative role early in the life of a new cell is part of a feedback loop whose pro-proliferation member is the ORC1-related protein CDC6, another important DNA replication factor.
“If cells integrate signals from their environment that promote another round of cell division, a pathway is engaged, in which a complex formed by Cyclin D and CDK4 (a cyclin-dependent protein kinase) triggers a cascade of effects that culminate in amplification of the cell’s commitment to cell division,” Hossain explains. The mechanism behind this amplification involves an interaction between Cyclin E-CDK2 and CDC6. Their level in the cell, in turn, is regulated by a transcription factor called E2F, which itself is controlled by a tumor suppressor protein called RB and an enzyme that adds methyl groups to histone proteins, called SUV39H1.
Hossain and Stillman report that early in the newly born cells, ORC1 is present at the promoter element of the gene that encodes Cyclin E, at which position it interacts with the RB protein and SUV39H1. The interaction results in repression of E2F-dependent transcription of the Cyclin E-encoding gene. Later in the G1 phase of the cell-division cycle, as cells receive signals that they should proliferate, CDC6 cooperates with Cyclin E and its protein kinase partner CDK2, to biochemically counter this repression, thus acting to dramatically increase the expression of the gene that encodes Cyclin E. This amplification process contributes to committing the cell to a new round of DNA duplication and chromosome segregation. Thus a feedback loop is established in which one DNA replication protein, CDC6, antagonizes the repressive action of another DNA replication protein, ORC1.
Many cancer cells overexpress Cyclin E and CDC6, and with others, the Stillman lab has shown previously that overexpression of Cyclin E causes genetic damage in the genome. “The opposing effects of ORC1 and CDC6 in controlling the level of Cyclin E thus contributes to the stability of the genome,” Stillman says. “It is a mechanism for directly linking the process of DNA replication with a cell’s commitment to divide.”
Written by: Peter Tarr, Senior Science Writer | publicaffairs@cshl.edu | 516-367-8455
Funding
The research described in this release was supported by the National Cancer Institute (CA13106) and an NCI Cancer Center Grant (CA45508).
Citation
“Opposing roles for DNA replication initiator proteins ORC1 and CDC6 in control of Cyclin E gene transcription” appears online in eLife on July 22, 2016. The authors are: Manzar Hossain and Bruce Stillman. The paper can be obtained at: DOI: http://dx.doi.org/10.7554/eLife.12785
Principal Investigator

Bruce Stillman
President and Chief Executive Officer
Oliver R. Grace Professor
Cancer Center Member
Ph.D., Australian National University, 1979