There is currently no cure for amyotrophic lateral sclerosis (ALS). But new research could open up avenues for better diagnosis and more effective treatment.
The study, led by Cold Spring Harbor Laboratory Associate Professor Molly Gale Hammell, analyzed the role proteins called TDP-43 play in ALS, also known as Lou Gehrig’s disease. In ALS, those proteins collect in nerve cells in the brain and spinal cord, causing sloughs of them to die off. This sweeping effect slowly paralyzes the patients.
Hammell’s lab, working closely with researchers from other leading institutions, recently published the findings in Cell Reports.
Hammell’s team tried to determine whether there were different types of ALS patients by studying the functions of the TDP-43 protein and its links to retrotransposons, also known as “jumping genes,” an important part of this genetic puzzle. The genes can randomly move from one spot of a chromosome to another. They can also alter gene expression, a process in which the instructions from DNA are translated into functional activities in the cell.
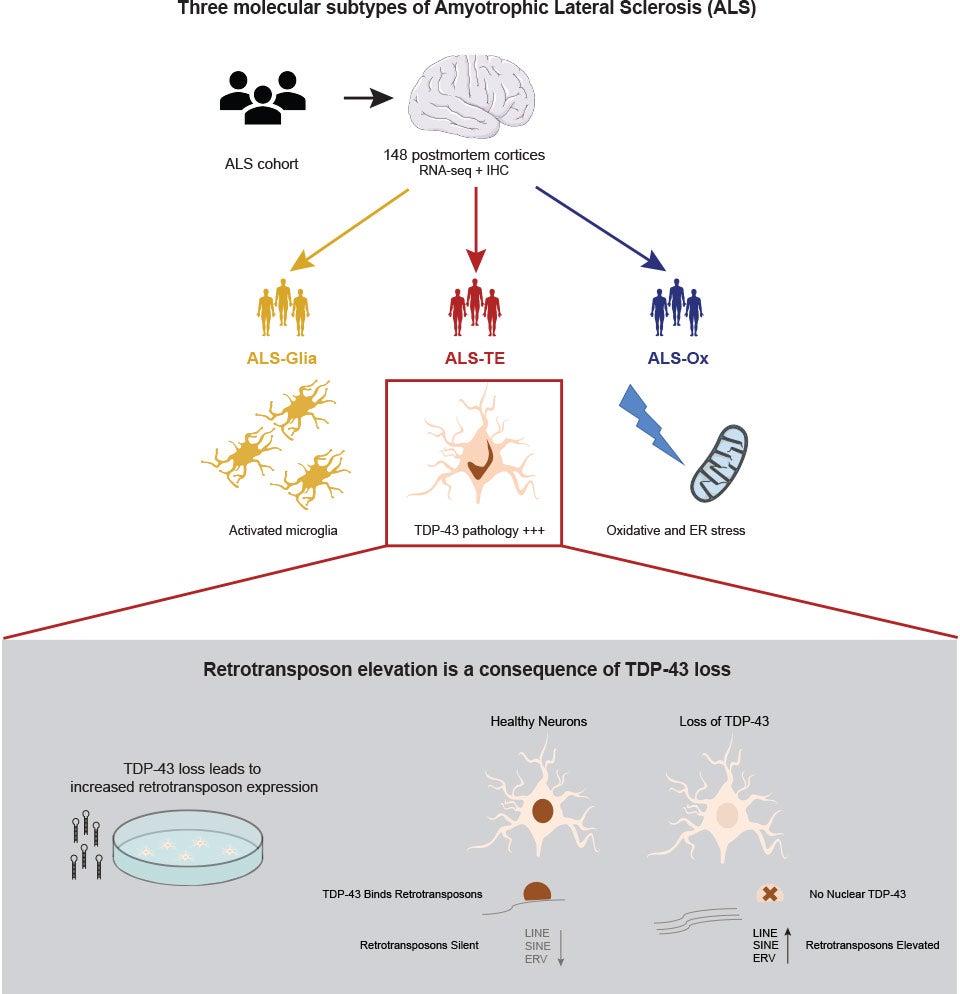
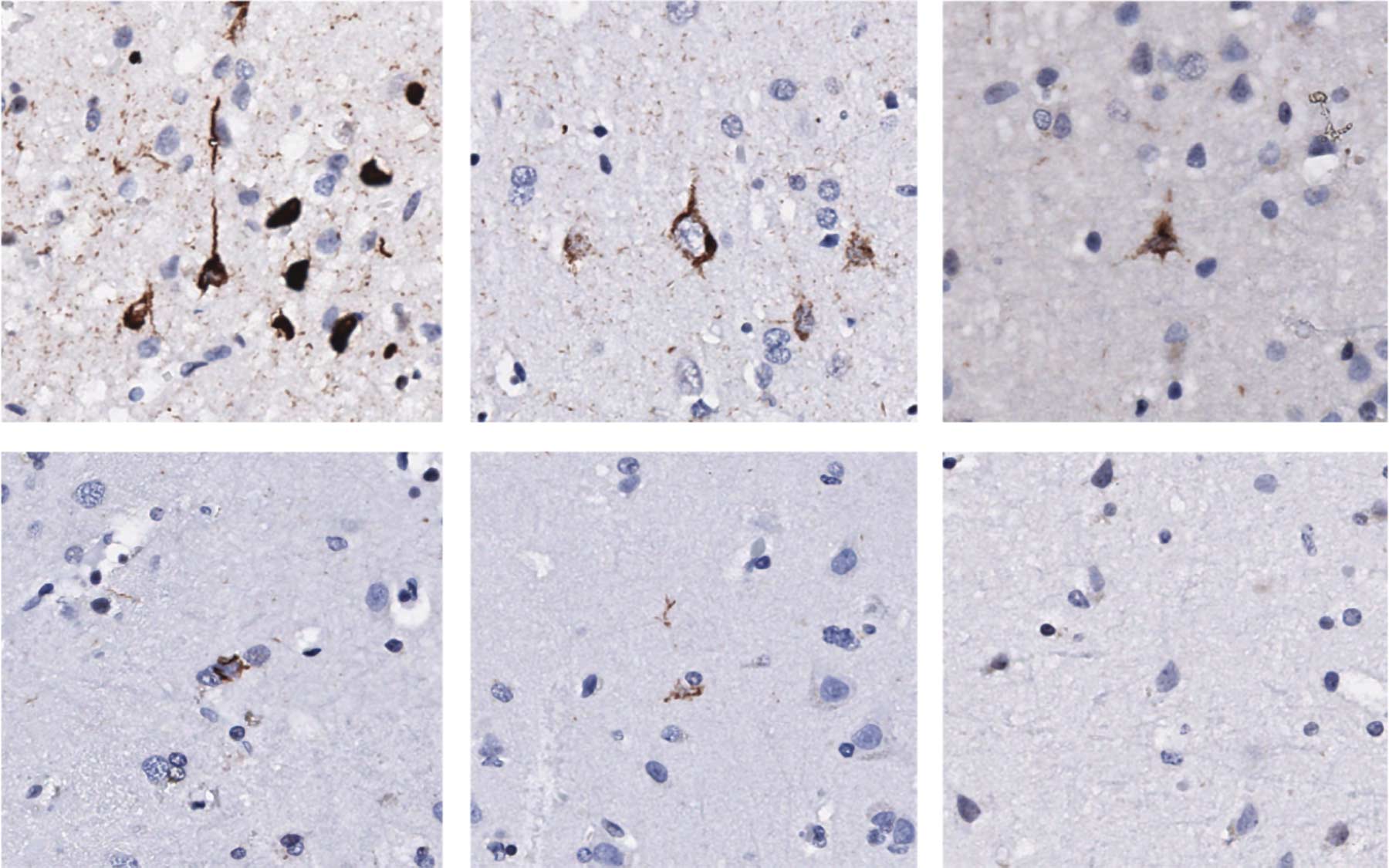
TDP-43 is one of the proteins that keep jumping genes silent. But when TDP-43 accumulates in clumps or aggregates in the nerve cells of ALS patients, it fails to silence the jumping genes. Hammell’s group found these telltale signs of TDP-43 pathology, or abnormal characteristics, and elevated transposons in some ALS patient samples.
“These jumping genes are telling us about patients who have TDP-43 pathology,” Hammell said. “We really don’t know why one patient would have one set of symptoms versus another, and we’re trying to answer that question.”
To find some of those answers, the team studied gene expression patterns in the brain tissues of post-mortem patients with and without TDP-43 pathology. They saw there was a de-silencing of jumping genes for the subset of patients with the most extensive TDP-43 pathology.
The team now wants to confirm whether the jumping genes actually contribute to the toxicity of cells in ALS patients. The team speculates that high levels of these jumping genes might mimic a viral pathogen or other infection in the tissues. If so, researchers may be able to directly target those genes with antiviral agents or other therapies.
“That’s one of the things we’re really excited about as a possibility,” Hammell said. “Going forward, we want to understand whether transposons are causing the disease, and whether they’re involved in other diseases that also tend to have TDP-43 aggregates, like frontotemporal dementia and some subset of Alzheimer’s.”
Researchers from Stony Brook University, Johns Hopkins University, Georgetown University Medical Center, The NYGC ALS Consortium, New York Genome Center, and University of California, San Diego, contributed significantly to the research and paper.
Written by: Charlotte Hu, Content Developer/Communicator | publicaffairs@cshl.edu | 516-367-8455
Funding
This research was funded by the Chan Zuckerberg Initiative, the Ride For Life Foundation, the Rita Allen Foundation of which Molly Hammell is a scholar, the O’Neil Charitable Trust, NIH/NINDS, NIH/NIA, the ALS Association, and the Tow Foundation.
Citation
Tam et. al, “Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia” appeared in Cell Reports on October 29, 2019.
Principal Investigator

Molly Gale Hammell
Associate Professor
Cancer Center Member
Ph.D., Dartmouth College, 2003
One thought on “Seeking better treatment for ALS, Lou Gehrig’s disease”
Comments are closed.