
Watson School graduate Lisa Krug studied ALS in flies, but the human element of this research always remained present for her. She explains in this guest blog post.
ALS is a disease that makes itself nearly invisible. Although each year 6,400 Americans are diagnosed with Amyotrophic Lateral Sclerosis, often called ALS or Lou Gehrig’s disease, they suffer inside their homes on hospice care—and the disease’s strikingly fast progression means that they will nearly all have passed on within a few years.This disease I now study only entered my own sphere of consciousness when my mother was diagnosed with it when I was in college. She passed away the year before I began graduate school here at Cold Spring Harbor, after three and a half years of struggling that forced her to leave behind every last shred of the independence that was such an integral part of her personality.
ALS is a progressive and fatal neurodegenerative disease that takes away your ability to walk, to use your hands, and ultimately your ability to sit upright, speak, swallow, and breathe independently. The neurons that control muscle motility gradually die, and so far no one has been able to determine what, precisely, is killing them off. There is no known cure, and only one treatment available that may extend lifespan on the order of a few months.
About 90% of ALS cases occur in people with no family history of the illness and in the absence of mutations that predict onset of the disease. That means you can live your life up to age 40 or 50 without ever knowing that your ultimate fate will be to be confined to a wheelchair and ultimately immobilized in hospice care with no therapeutic options.
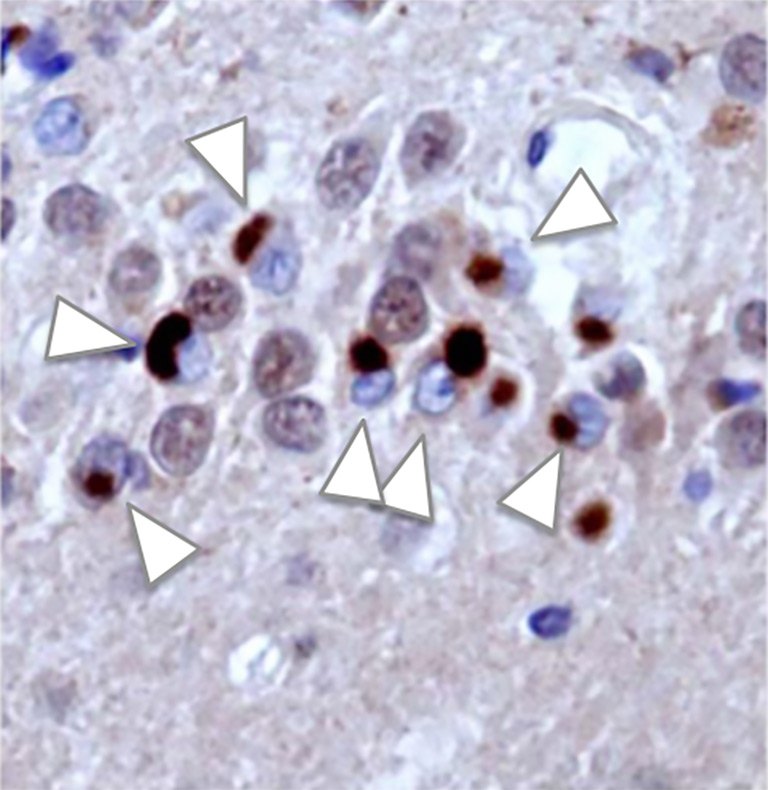
This is what makes a protein called TDP-43 so intriguing. In over 98 percent of ALS patients, ALS-affected neurons accumulate TDP-43 in an abnormal place in the cell. This easily identifiable pathology enabled me and my colleagues in the Dubnau lab to use TDP-43 to model ALS in lab animals. In the course of this research, we discovered a specific class of small RNAs—molecules that regulate gene expression within cells—whose function had not previously been known to be disrupted by TDP-43 pathology. The disruption we observe directly results in cell death in the nervous system, in a model of the disease we’ve created in fruit flies.
Using both genetic methods and drug treatments directed at the downstream targets of this class of small RNAs, thereby bypassing the disruption we observe in response to TDP-43 pathology, we are able to largely block both the nervous system cell death and the dramatically shortened lifespan of our flies compared to untreated animals. These findings present a direct new avenue for possible therapies that could halt motor neuron cell death and possibly provide a much-needed set of diagnostic tools for early detection in human patients. We consider our work, though preliminary, an important step forward—a ray of hope for the often-overlooked community of ALS sufferers and their loved ones whose outlook is otherwise so bleak.
More common neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases have historically relegated ALS to the shadows. But the ALS cause has recently begun to benefit from increased awareness, fundraising, and research. Both the Dubnau lab and our partners in Dr. Molly Hammell’s lab at CSHL are proud recipients of research funding from Ride for Life, a remarkable not-for-profit organization directed by Dr. Chris Pendergast—an ALS sufferer—and his wife.
MyLITV report from the 2016 Ride for Life stop at CSHL
The organization raises money for ALS research via their annual Ride for Life, in which ALS patients and their friends and family ride their wheelchairs from the eastern end of Long Island into Manhattan across the Brooklyn Bridge every May, stopping along the way to raise both money and awareness for the disease.
Members of the Dubnau lab and I walked with Ride for Life and hosted an event here at CSHL last May. The Pendergasts’ work is truly inspiring. It educates those who have no awareness of the disease, and provides both support and a constructive outlet for those who have been directly diagnosed or otherwise touched by ALS. It is the kind of resource I wish our family had when my mother was diagnosed several years ago and brings a very human element to the scientific research that we do—shedding light on this nearly invisible but devastating disease.
More information about ALS, multiple assistance and support programs for ALS patients and their caretakers and loved ones, as well as information on how to donate and participate in the annual Ride can be found on the Ride for Life website: www.ride-for-life.org