Truncated p53 proteins, presumed unimportant, now point to new drug targets for some of ‘the hardest cancers’
Cold Spring Harbor, NY — Discovered in the 1970s, tumor suppressors are among the most important proteins in the body. A master regulator of growth—“the guardian of the genome”—the p53 protein monitors cell growth for errors. We rely on suppressors like p53 to defeat cancer before it takes root. Indeed, cancer cells cannot survive unless p53 is mutated or non-functioning.
Not surprisingly, the gene that encodes p53 is the most frequently mutated gene found in human cancers. Most p53 gene mutations prevent p53 from being functional. In new research appearing in eLife, scientists at Cold Spring Harbor Laboratory (CSHL) and Memorial Sloan Kettering Cancer Center (MSK) focused on certain “loss of function” p53 mutations, and found that the mutant proteins they generate can act to promote cancer growth and metastasis, not halt it.
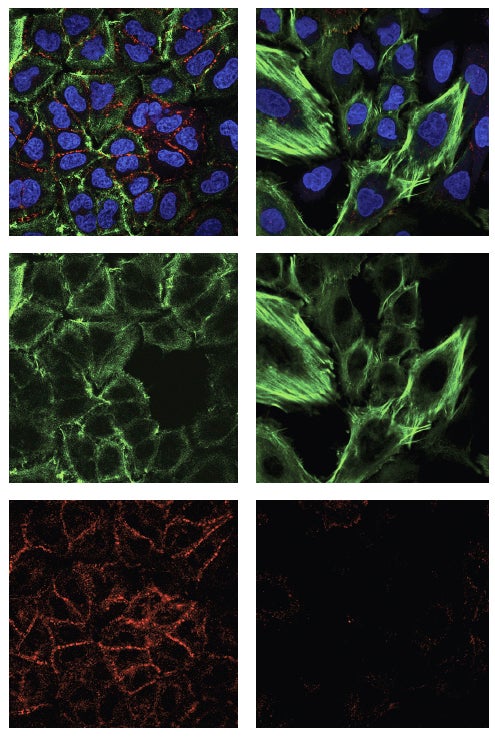
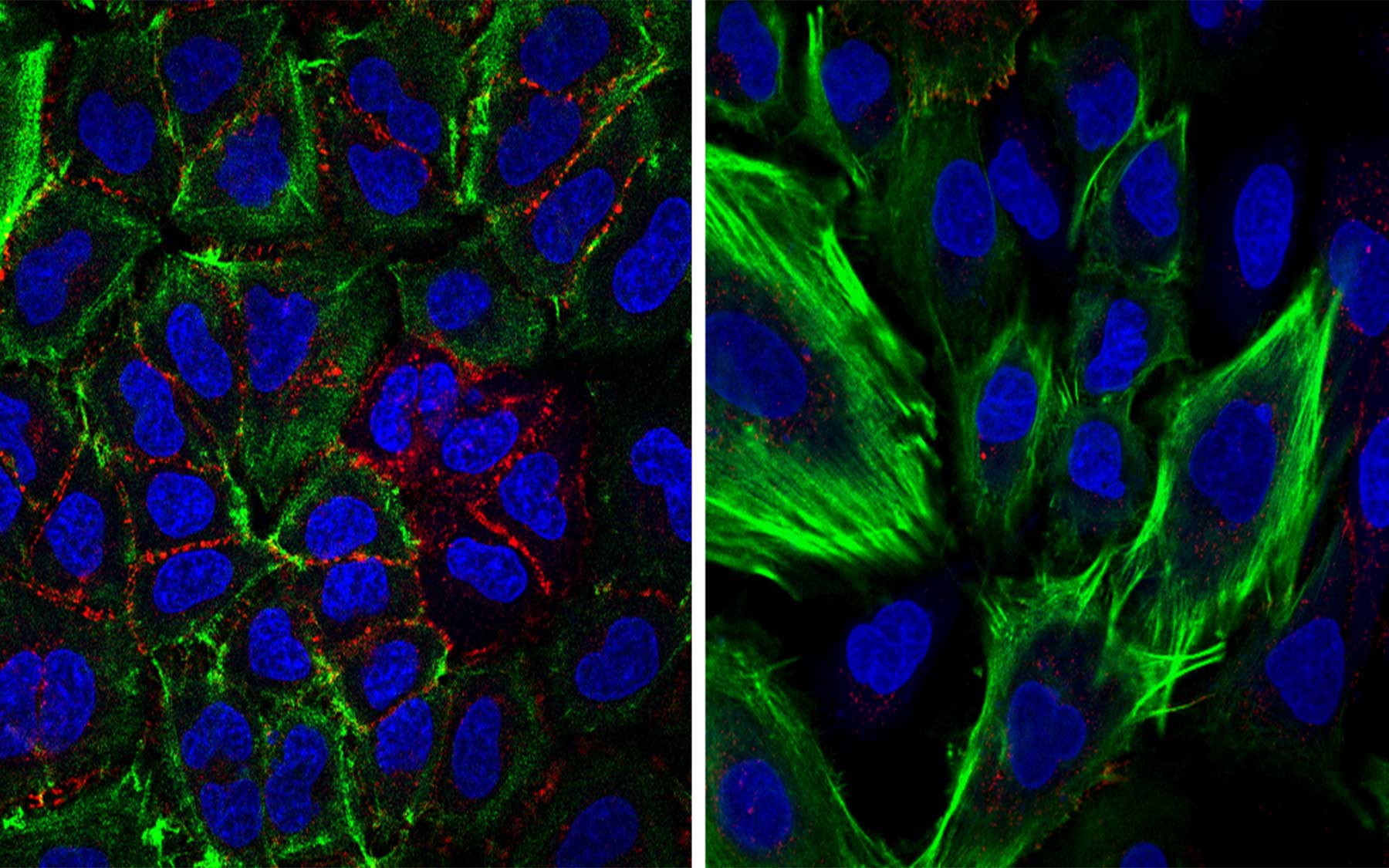
The variants studied are truncated—“they lack roughly half of the domains, or units, of the full-length p53 protein, specifically the domains that enable full-length p53 to enter the cell nucleus and bind DNA, both essential in its normal tumor-suppressor function,” says CSHL Associate Professor Raffaella Sordella, who led the research in a collaboration with Professor Scott Lowe, a former CSHL colleague now at Memorial Sloan Kettering.
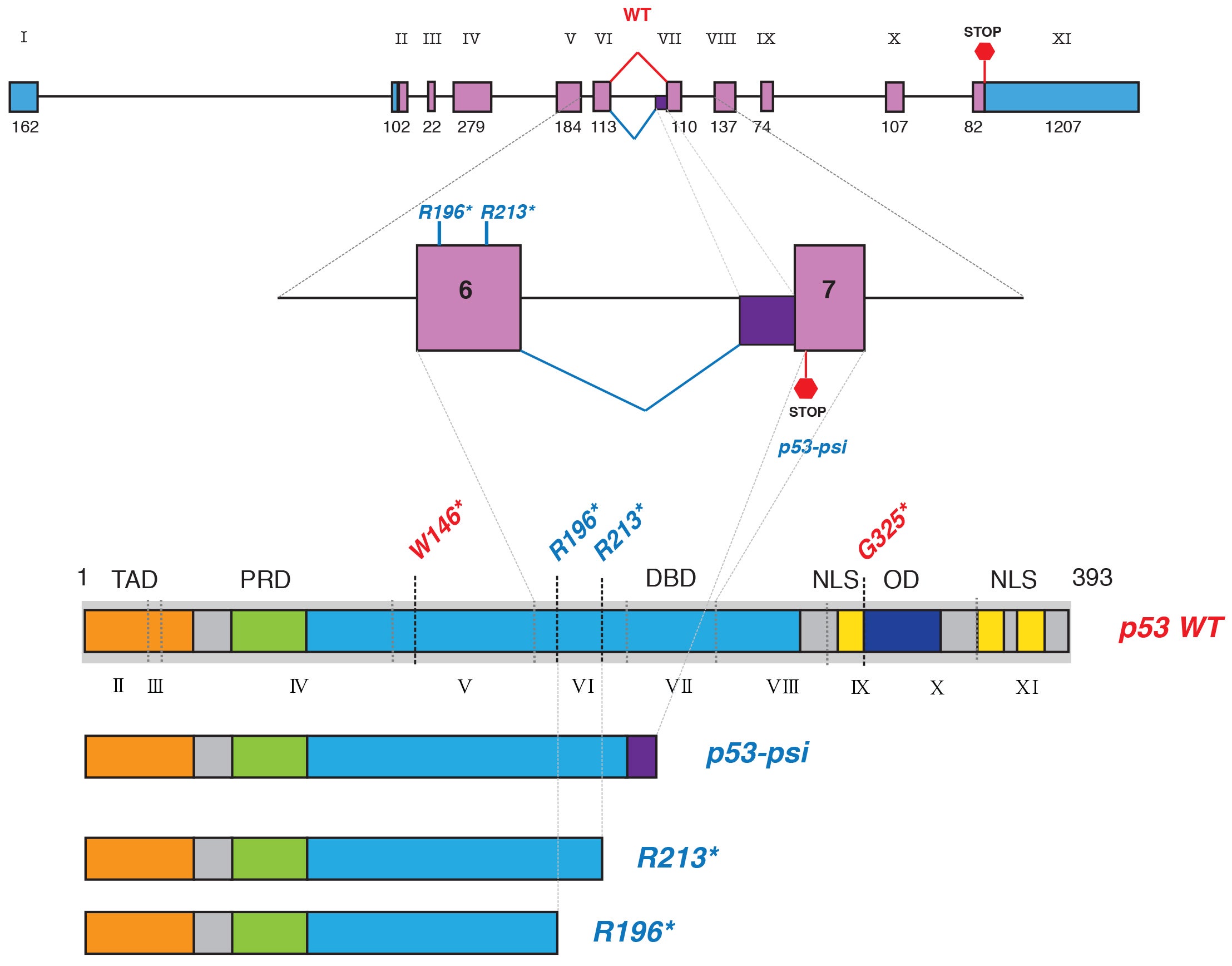
Until the CSHL-led research, the studied variants were presumed unimportant. In studies in cell culture and living mice, the team performed an array of experiments demonstrating that p53 proteins truncated after the 6th protein-coding segment, called exon 6, “no longer function as tumor suppressors but instead promote cancer by directly altering the functions of mitochondria,” according to Sordella. Mitochondria are the tiny energy factories found in great abundance throughout the cell’s cytoplasm.
By studying data on human tumors, the team learned that the truncated p53 proteins were most likely to be found in what Sordella calls “the hardest cancers”—ones that resist treatment and are likely to reappear even if surgically removed. Indeed, abundance of exon 6-truncated p53 proteins seems to predict bad outcomes even in cancers treated early and aggressively.
The studied p53 mutations are similar to another truncated version of p53, discovered by Sordella’s team in 2014. Called p53Ψ (the Greek letter “psi”), this mutated form of p53 is also missing domains that enable it to enter the nucleus and bind DNA. Instead, it localizes to the mitochondria and promotes tumor progression and metastasis. Unlike the exon 6-truncated p53 protein, however, p53Ψ is the result not of a DNA mutation but rather an alternatively spliced RNA copy of the p53 gene’s DNA message.
The similarities observed by Sordella’s team led them to explore whether the cancer-promoting impact of the two truncated p53s could be traced to the same mechanism. The team’s experiments revealed that it was.
Like p53Ψ, exon 6-truncated p53 appears to promote cancer by locating to mitochondria, where it docks with a membrane protein called Cyclophilin D (CypD). The latter regulates the opening of tiny pores in the mitochondria membrane. When exon 6-truncated p53 or p53Ψ interact with CypD, the inner mitochondria pores open only for a brief time, having the effect, Sordella believes, of altering mitochondrial functions. And this, she further suspects, is what leads to the cancer-promoting effect that both truncated versions of p53 seem to engender. In fact, the team found that cancer cells harboring mutations that lead to the production of the exon 6-truncated p53 are dependent on p53 and CypD.
“Remarkably, despite 40 years of research and over 80,000 publications on p53, our new findings show that it still holds mystery and promise,” Sordella says.
“It seems that by changing mitochondrial function, the variants are priming cells to reprogram themselves,” Sordella adds. Specifically, cells that are normally epithelial—for instance, cells that line the lung or pancreas—undergo a change of type, taking on the characteristics of mesenchymal cells. Such cells are liable to break loose from their moorings in tissue and move via the circulation to other parts of the body. This is thought to be a prime enabling factor in cancer metastasis.
Sordella says the team’s findings could lead to improved cancer therapies. “These mutations are strong candidates for targeting by precision medicine. The frequency of exon-6 truncating mutations in fact is comparable to other precision medicine targets such as the EGFR oncogenic mutations found in lung cancer. We have begun discussing with several pharmaceutical companies ways in which we can use our newly gained knowledge to develop treatments that will make a positive difference for many cancer patients.”
Sordella and her collaborators plan to take the new findings from the bench to patients. “It is our goal to screen patients for the presence of these mutations and treat with p53 Ψ signaling inhibitors, such as small molecules inhibiting CypD, according to their tumor genotype. Our hope is that this will extend and perhaps save lives.”
Written by: Peter Tarr, Senior Science Writer | publicaffairs@cshl.edu | 516-367-8455
Funding
The research discussed here was supported by the National Cancer Institute (NCI P01 CA129243-06 target for therapy for 652 carcinomas in the lung) and Swim Across America.
Citation
“TP53 exon-6 truncating mutations produce separation of function isoforms with pro-tumorigenic functions” appears online in eLife. The authors are: Nitin H Shirole, Debjani Pal, Edward R Kastenhuber, Serif Senturk, Joseph Boroda, Paola Pisterzi, Madison Miller, Gustavo Munoz, Marko Anderluh, Marc Ladanyi, Scott W Lowe and Raffaella Sordella. The paper can be accessed at: https://elifesciences.org/content/5/e17929