
The disease, hereditary spastic paraparesis (HSP), is actually a heterogeneous group of genetic disorders whose primary symptoms are progressive spasticity and weakness in leg and hip muscles. By the time they were two years old, the patients in the study—three brothers now aged 20, 15 and 14—developed stiff legs and abnormal gait that progressively worsened until they could no longer walk long distances or play soccer by the age of 13.
HSP, which is thought to affect roughly 20,000 people in the United States, is estimated to exist in about 30 forms. All are monogenic in origin, i.e., caused by a mutation in a single gene. Scientists have so far identified 20 HSP-related genes. To find the exact genetic mutation behind the boys’ condition, Dr. Orly Elpeleg’s group at the Hadassah Medical Center in Jerusalem and her CSHL collaborators led by Dr. Yaniv Erlich, then a graduate student in Dr. Greg Hannon’s lab at CSHL and currently a Fellow at the Whitehead Institute, started with a relative new approach called exome sequencing.

Instead of sequencing the entire genome, scientists use this method to focus on just the exome—the total set of “exons,” or genomic regions that code for protein (about 2% of the genome). In a previous collaboration with Elpeleg, Erlich and Hannon used exome sequencing to rapidly identify the mutated gene that causes another devastating inherited neurological disease called Joubert Syndrome.
“With exome sequencing, which has been used more and more in the last two years, we can, in a rapid, cost-effective way, focus on high-value areas in the genome and boost ‘coverage’ of those areas to more confidently and statistically identify the gene variants associated with rare Mendelian disorders,” explains Dr. Emily Hodges, a research investigator in the Hannon lab who developed some of the technological aspects of this approach.
“But the downstream part of the package is still lacking, and that’s the informatics strategy that’s needed to filter out most of the mutations that we find and zero in on the disease-causing one.” That’s the part that’s been enormously improved by this current study, according to Hodges.

When a comparative analysis of the boys’ and their parents’ exomes, which Hodges helped sequence, suggested that the causative mutation wasn’t within any of the 20 known HSP-related genes, the team hunted for the unknown gene using homozygosity mapping. This is a method used to narrow down the genomic location of genes that cause disease in a recessive way—i.e., when both copies of the gene are mutated—in patients who are from families with a history of marriage between kinsmen.
The final step was to sift through the candidate genes via “disease-network analysis” and filter out those genes that did not “functionally behave” like HSP-related genes. “By integrating what was previously known about the disease’s molecular signature through studies of closely related disorders and then ruling out non-causative or bystander genetic variations, disease-network analysis adds another layer of information to the data that we get from sequencing,” explains Erlich.
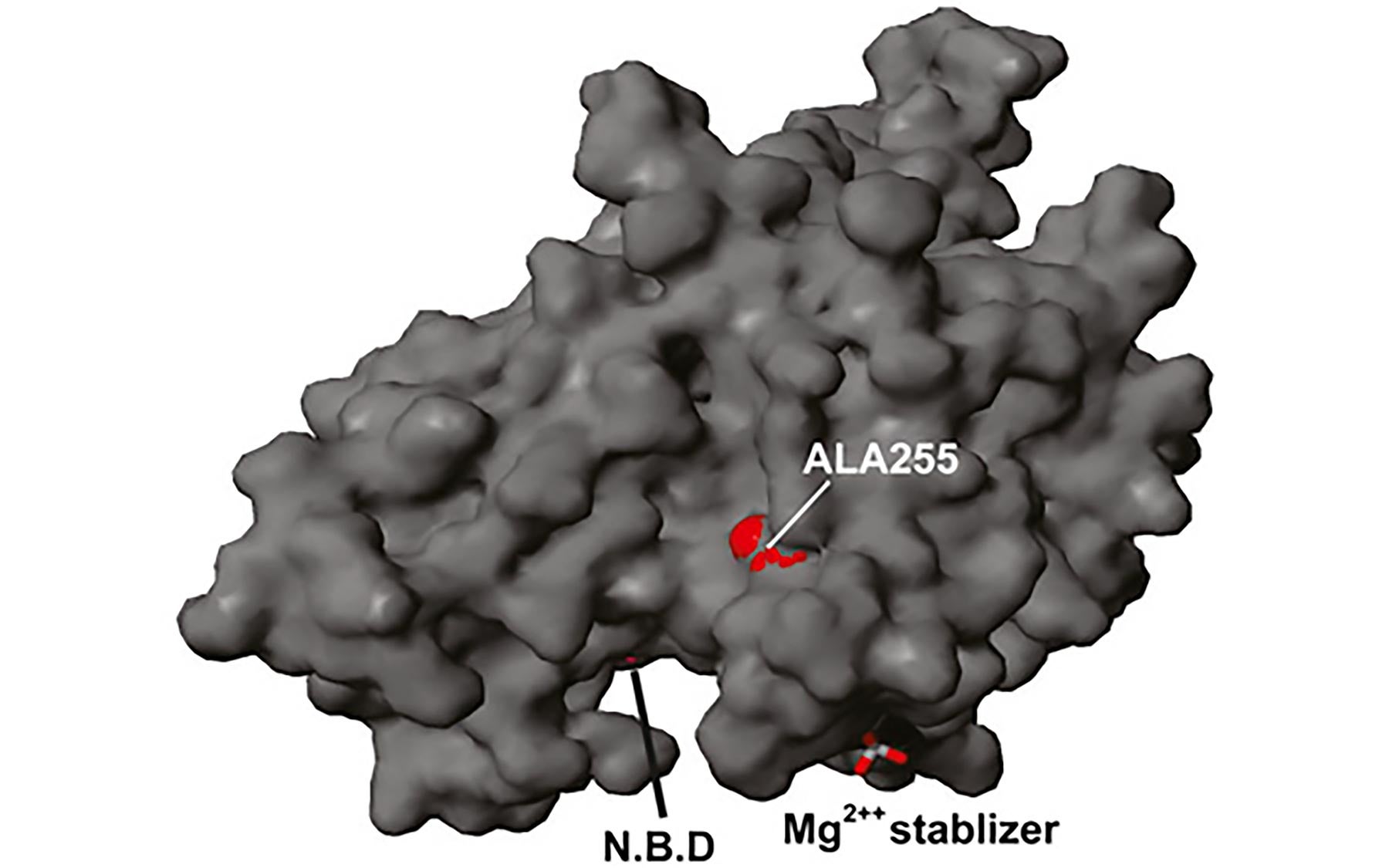
This three-step strategy led the team to a gene called KIF1A and the mutation within it that caused the boys’ disease. The KIF1A gene encodes a protein called kinesin, a molecular motor that transports cargo from one point to another inside cells. It’s especially crucial for the survival of our bodies’ nerve cells, in which cargo has to be transported though long axonal cables from the cell body to the nerve endings. The scientists have found that 1 in 200 Palestinians carry this KIF1A mutation.
With this strategy, the scientists now plan to hunt for the genetic variants responsible for 200 other rare familial single-gene disorders. The information they uncover can be used to screen individuals for disease-causing mutations so that asymptomatic carriers will be able to avoid unknowingly passing the condition to their descendants.