Tested on a newly discovered target and candidate drug for leukemia, the method has broader application
Cold Spring Harbor, NY — A collaborative effort by cancer researchers at Cold Spring Harbor Laboratory (CSHL) and chemists at Boehringer Ingelheim (BI), a pharmaceutical firm, has resulted in the identification of a new drug target in leukemia and creation of a candidate drug that hits the target. Perhaps even more important, the research demonstrates a new, highly accurate way of proving how this and certain other classes of drugs work—extremely valuable information in the risky business of drug development. The research appears today in Nature Chemical Biology.
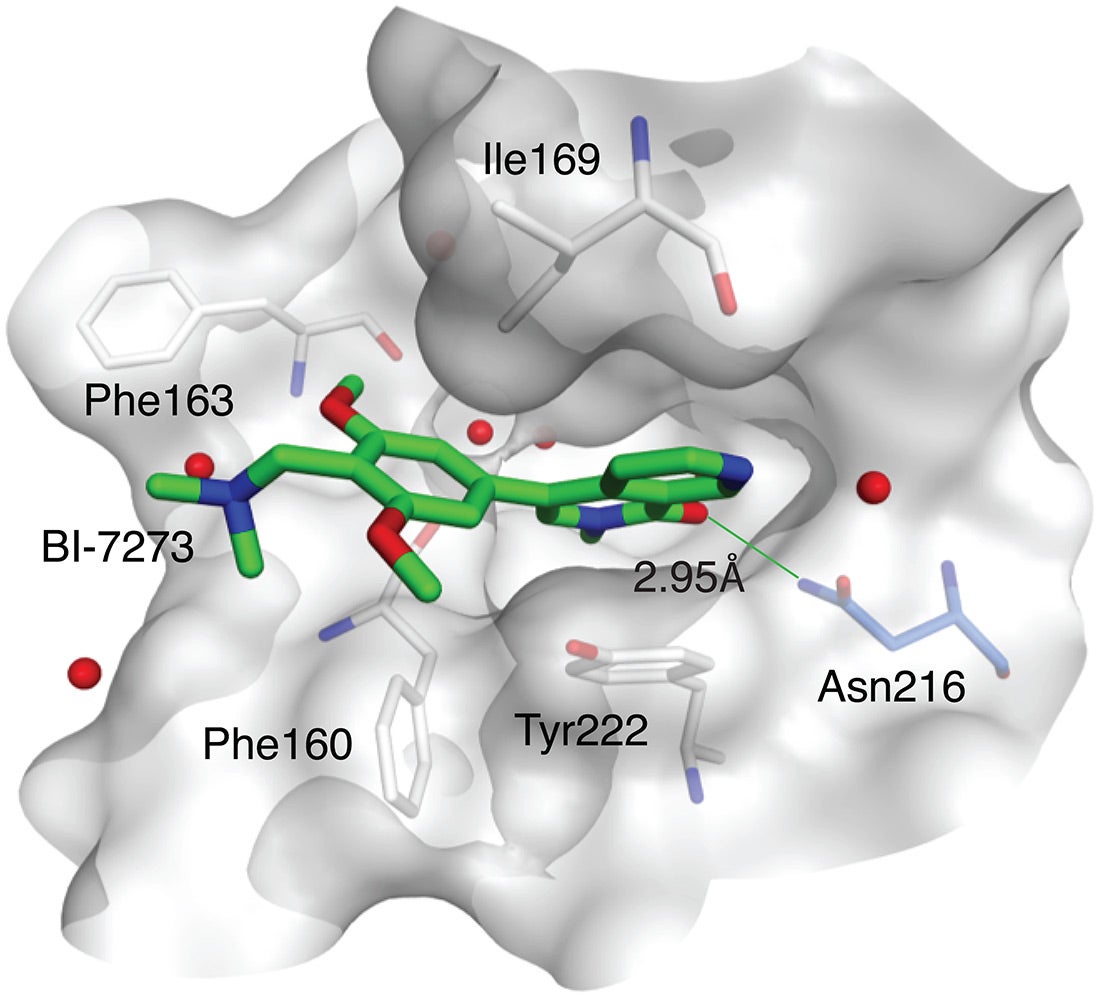
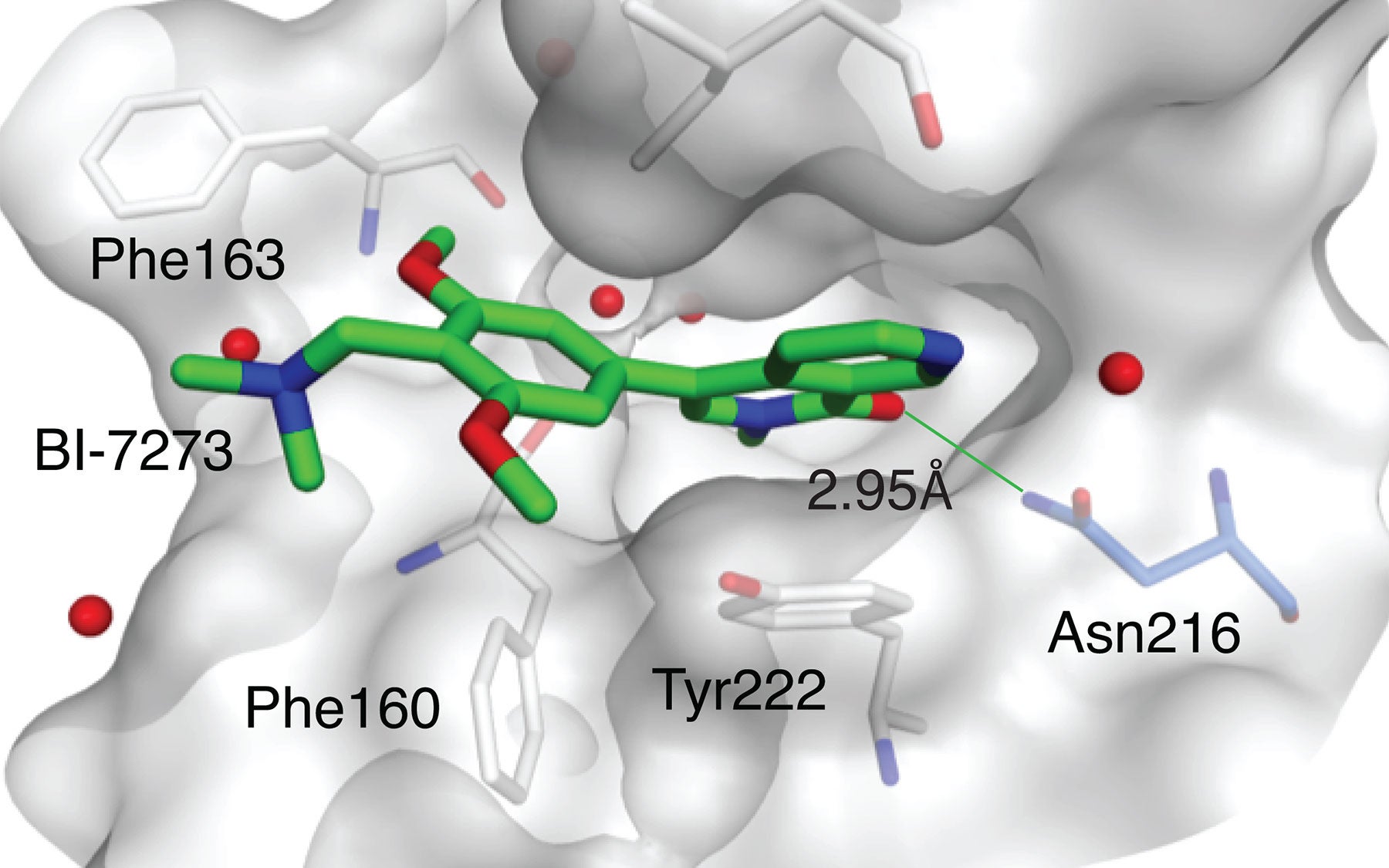
The cancer in question is a particularly aggressive form of blood cancer called acute myeloid leukemia, or AML, which is often fatal. The newly identified target is a binding pocket on a protein called BRD9. The drug designed by BI to bind at that pocket, called a bromodomain, is provisionally called BI-7273.
The team of academic and industry scientists, led by Associate Professor Christopher Vakoc of CSHL in coordination with Dr. Manfred Koegl of BI, identified BRD9 in screens, then demonstrated its importance in human AML cells grown in culture and in rodent models of AML.
Like nearly all proteins, BRD9 is subdivided into sections called domains, each of which has a specific function. Each domain is encoded by a portion of the BRD9 gene. The domain of interest to Dr. Anja Hohmann, first author of the new paper who performed the research while still a graduate student in Vakoc’s lab, was BRD9’s bromodomain, the subunit of the protein that “reads” chromatin, a bundled form of the cell’s genetic material.
Experiments led by Hohmann demonstrated that BRD9 is a “dependency” of AML: the human cancer depends on the protein in order to remain “immortal,” i.e., capable of proliferating rapidly, unimpeded at the usual checkpoints that cells have evolved to limit growth. Specifically, the team discovered BRD9’s action was necessary for activation of the cancer gene MYC, a master orchestrator of signaling pathways that enable cells to grow uncontrollably.
BI scientists developed several candidate molecules that could bind at the BRD9 protein’s bromodomain. This work led to identification of the compound dubbed BI-7273, which, when tested against AML cells, selectively limited proliferation of the cancer cells, and prevented activation of the MYC oncogene.
“We were of course pleased with these results,” Vakoc says. “But we set an even higher bar. We wanted to be able to show, unambiguously, how the drug worked—we wanted to prove that its target in AML cells was the bromodomain of the BRD9 protein.” The team’s working hypothesis was that this bromodomain—which forms a deep pocket on BRD9’s surface—is the binding site of the drug BI-7273.
There are various ways of proving that a binding site’s interaction with a drug is the direct cause of the drug’s effect. The most commonly used method is to change the shape or chemical characteristics of the pocket—the binding site, in this case BRD9’s bromodomain—so that the candidate drug can no longer attach there.
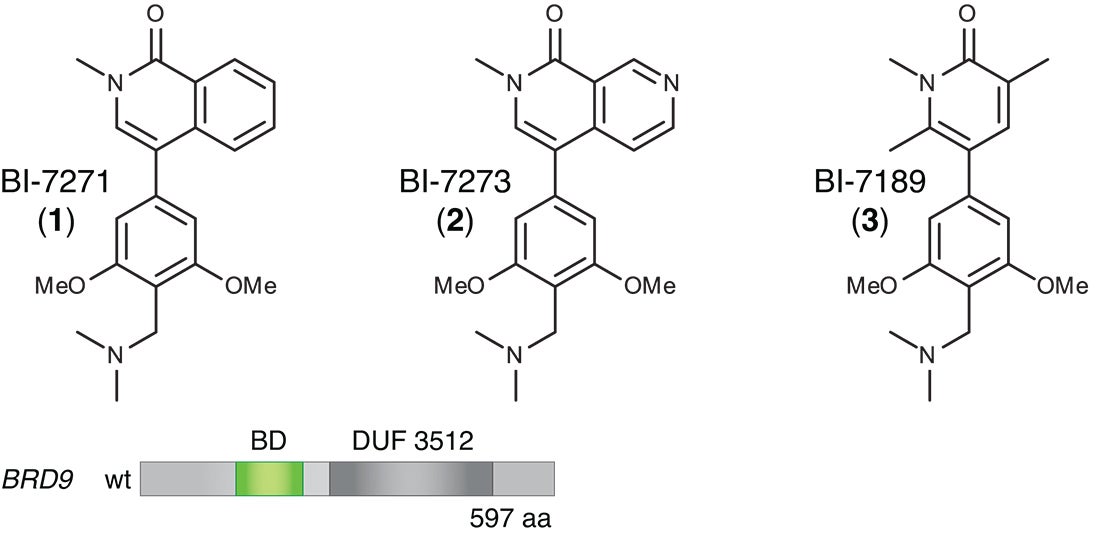
(bottom) Schematic of the BRD9 protein’s domain structure. The protein has multiple subunits, one of which is a bromodomain (BD, colored green) specific to BRD9. Hohmann and Vakoc swapped this particular domain for the bromodomain of another, similar protein, BRD4. The modified version of BRD9 (with BRD4’s bromodomain swapped in) continued to perform an essential function in enabling AML cells to proliferate. This modified protein was “resistant” to the action of BI-7273, proving that the drug’s target is the bromodomain native to BRD9.
Changing the shape of the binding pocket has been likened to changing the lock on a door so that a specific key, i.e., the drug, can no longer fit it. A method called structure-guided mutagenesis is typically used to do this. It is intricate, involving determination of the precise atomic configuration of the binding pocket and knowledge of how the drug, or “key,” fits into it.
“We figured out a much easier and equally rigorous way of doing this,” Hohmann says, “that exploits the modularity of proteins.” There are 61 known bromodomain “motifs” in at least 48 different proteins, of which BRD9 is one. Using simple copy-and-paste techniques, she and the team “swapped out” the bromodomain of the BRD9 protein and replaced it with the bromodomain that’s part of a protein called BRD4.
The team was surprised that the now-modified BRD9 protein continued to function in AML cells. This may be due to a “family resemblance” in bromodomains—while the bromodomains of BRD9 and BRD4 are different at the atomic level, they are, says Vakoc, “functionally synonymous.” The modified version of BRD9 continued to facilitate the reading of chromatin, and AML cells continued to depend on its action to engage signaling pathways activated by MYC. Cancer cells with the modified BRD9 continued to grow out of control.
This provided a perfect test bed for BI’s new drug, BI-7273. In AML cells with the “swapped out” BRD9 bromodomain, the drug, as hoped, had no impact on AML proliferation.
“This proved that it was specifically the ‘native’ bromodomain of BRD9 that accounted for the drug’s anti-cancer activity. When we swapped it out, even though the protein continued to function, the drug could no longer attach to it. It’s a very elegant way of showing precisely how the drug had its desired effect, by binding the bromodomain native to BRD9,” Vakoc says.
In other experiments, the team showed the “domain-swapping” technique is “generalizable” in other situations where functionally synonymous domains can be swapped in for domains native to a protein that are thought to be key to a drug’s action. In AML cells, they swapped a domain of the protein EZH2 with a corresponding and functionally synonymous domain from EZH1. Doing so prevented a candidate drug called GSK126, thought to be effective by interacting with wild type EZH2, from impeding the growth of AML cells.
“Drugs and their targets are not really like keys and locks,” Vakoc explains. “In many instances, a single key will fit many different locks. In other words, in many cases, a drug will interact with many proteins, and because of this we don’t know its precise target. Off-target binding is frequently a cause of unwanted side effects.
“As the age of precision medicine begins, this is an important issue, a matter of sink or swim for some candidate drugs. Here we’ve described a simple new approach that can unambiguously assign the therapeutic effect of a drug to a single binding site.”
Written by: Peter Tarr, Senior Science Writer | publicaffairs@cshl.edu | 516-367-8455
Funding
The research described here was supported by grants from Cold Spring Harbor Laboratory NCI Cancer Center Support grant CA455087. Additional funding was provided by National Institutes of Health grant NCI RO1 CA174793, the Alex’s Lemonade Stand Foundation, the V Foundation, Pershing Square Sohn Cancer Research Alliance, and by Boehringer-Ingelheim. Individual investigators received support from the Martin Sass Foundation, the Lauri Strauss Leukemia Foundation, the Burroughs-Wellcome Fund Career Award.
Citation
“Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition” appears online in Nature Chemical Biology on July 5, 2016. The authors are: Anja F. Hohmann, Laetitia J. Martin, Jessica Minder, Jae-Seok Roe, Junwei Shi, Steffen Steurer, Gerd Bader, Darryl McConnell, Mark Pearson, Thomas Gerstberger, Teresa Gottschamel, Diane Thompson, Yutaka Suzuki, Manfred Koegl, and Christopher R. Vakoc. The paper can be accessed here: http://www.nature.com/nchembio/journal/vaop/ncurrent/full/nchembio.2115.html
Principal Investigator

Chris Vakoc
Professor
Alan and Edith Seligson Professor of Cancer Research
Cancer Center Deputy Director of Research
M.D., Ph.D., University of Pennsylvania, 2007